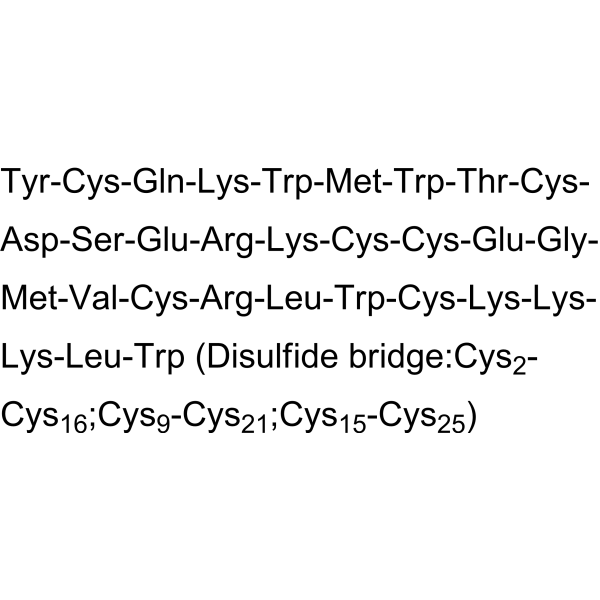Search Result
Results for "
subtype selectivity
" in MedChemExpress (MCE) Product Catalog:
1
Biochemical Assay Reagents
7
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-133012
-
|
|
TRP Channel
|
Neurological Disease
|
|
GFB-8438 is a potent and subtype selective TRPC5 inhibitor, with IC50s of 0.18 and 0.29 μM of hTRPC5 and hTRPC4, respectively. GFB-8438 shows excellent selectivity against TRPC6, other TRP family members, NaV 1.5, as well as limited activity against the hERG channel. GFB-8438 protects mouse podocytes from injury induced by protamine sulfate model .
|
-
-
- HY-103229
-
|
|
iGluR
|
Neurological Disease
|
|
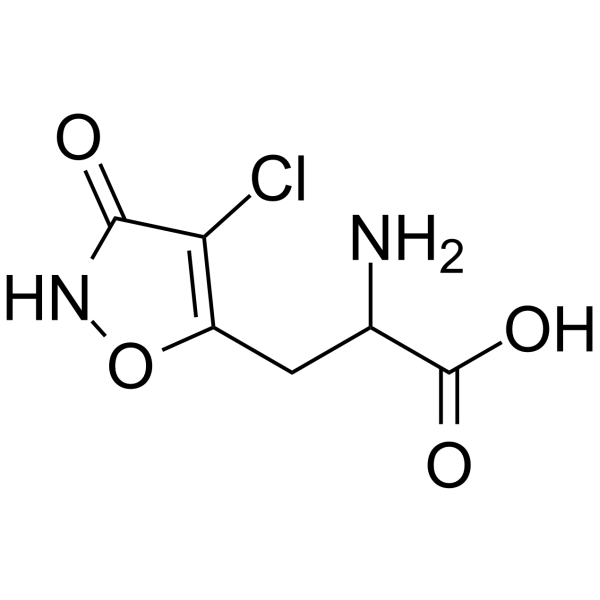
Cl-HIBO is a highly subtype-selective GluR1/2 agonist (EC50=4.7 and 1.7 μM, respectively). Cl-HIBO is a potent AMPA receptor agonist (IC50=0.22 μM). Cl-HIBO has desensitizing properties .
|
-
-
- HY-142723
-
|
|
Potassium Channel
|
Neurological Disease
|
|
KCa2 channel modulator 1 (compound 2o) is a potent subtype-selective positive modulator of KCa2 channel. KCa2 channel modulator 1 potentiates human KCa2.3 channels with an EC50 value of 0.19 μM and 0.99 μM on the rat KCa2.2 channel subtype .
|
-
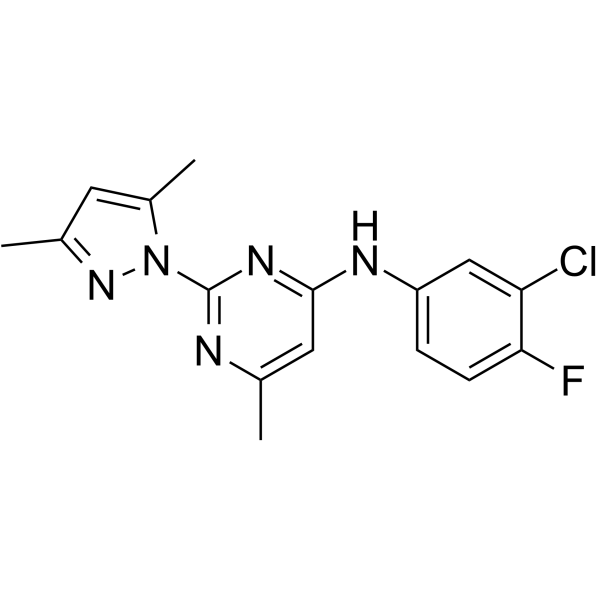
-
- HY-142735
-
|
|
Potassium Channel
|
Neurological Disease
|
|
KCa2 channel modulator 2 (compound 2q) is a potent subtype-selective positive modulator of KCa2 channel. KCa2 channel modulator 2 exhibits similar potency on the rat KCa2.2a and human KCa2.3 channel subtypes, with EC50s of 0.64 μM and 0.60 μM, respectively
|
-
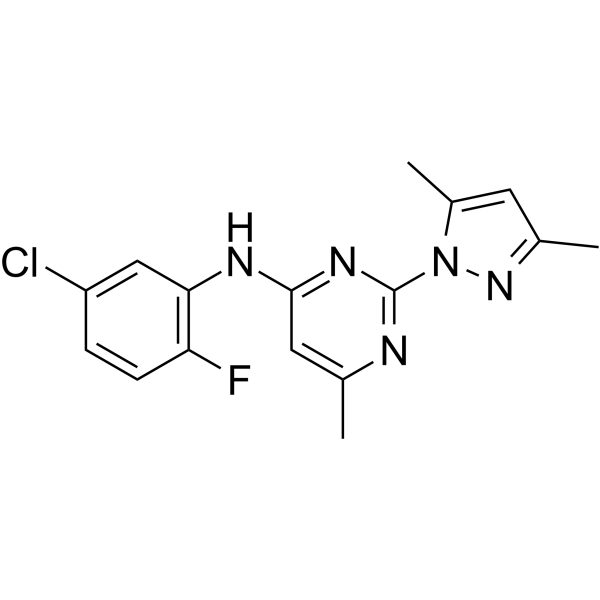
-
- HY-103066
-
|
|
nAChR
|
Neurological Disease
|
|
Br-PBTC is a potent, 2/4 subtype-selective positive allosteric modulator of nAChRs (nicotinic acetylcholine receptors) with α2β2,α2β4,α4β2,α4β4,(α4β2)2α4 and (α4β2)2β2 EC50 ranges from 0.1~0.6 μM. Br-PBTC acts from the c-tail of an α subunit .
|
-
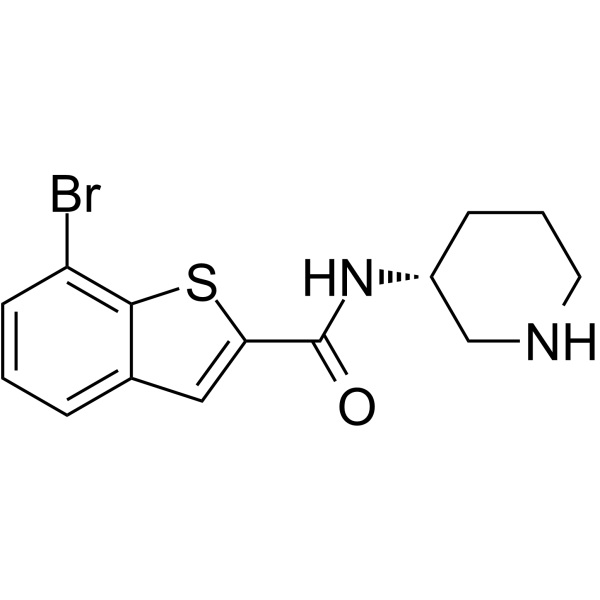
-
- HY-115764
-
-
-
- HY-B0374A
-
-
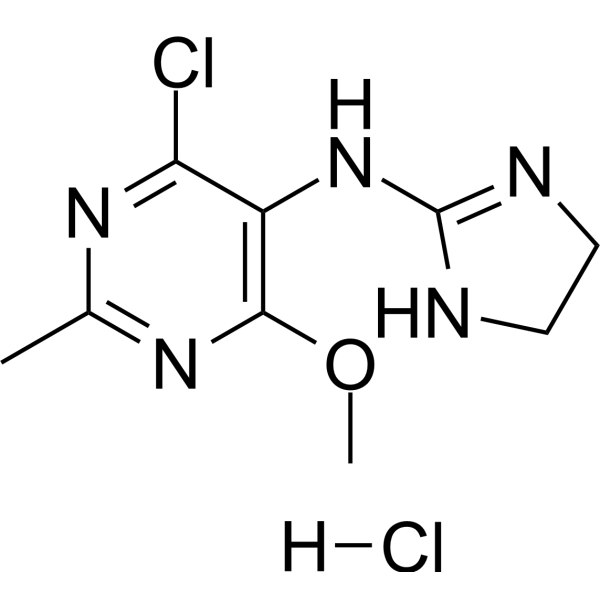
-
- HY-15831
-
-
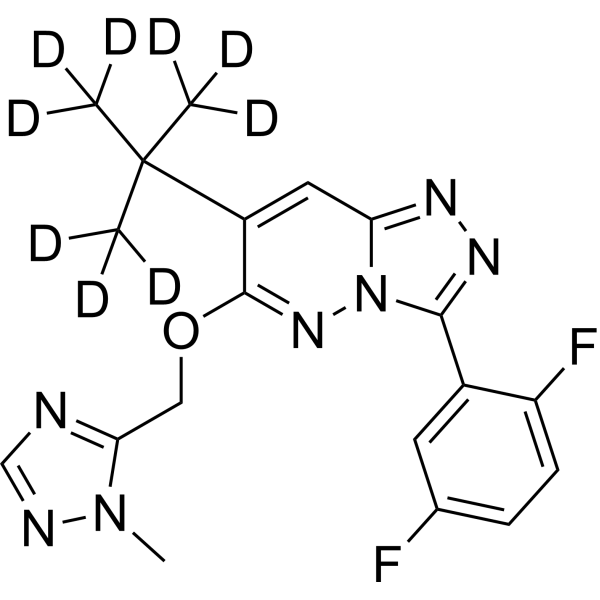
-
- HY-150692
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
Estrogen receptor α antagonist 1 (compound 35) is a highly selective antagonist of estrogen receptor α, with IC50s of 0.02, 6.55 and 7.73 μM for estrogen receptor α, estrogen receptor β and MCF-7 cells, respectively. Estrogen receptor α antagonist 1 can be used for the research of cancer .
|
-
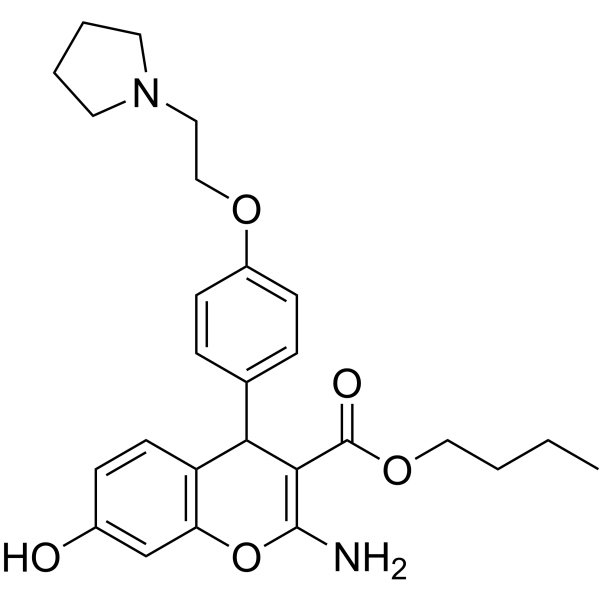
-
- HY-101308A
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
MRS2179 tetrasodium hydrate is a competitive P2Y1 receptor antagonist, with a Kb of 102 nM and a pA2 of 6.99 for turkey P2Y1 receptor. MRS2179 tetrasodium hydrate is selective for P2Y1 over P2X1 (IC50=1.15 µM), P2X3 (12.9 µM), P2X2, P2X4, P2Y2, P2Y4, and P2Y6 receptors . MRS2179 tetrasodium hydrate inhibits platelet aggregation .
|
-
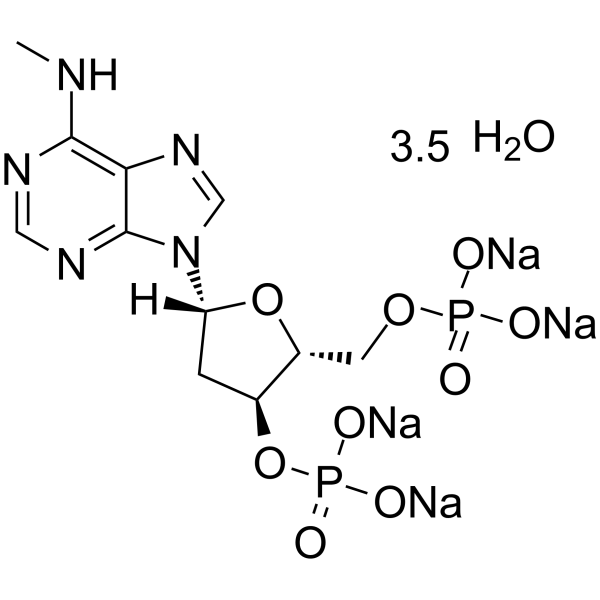
-
- HY-142868
-
-
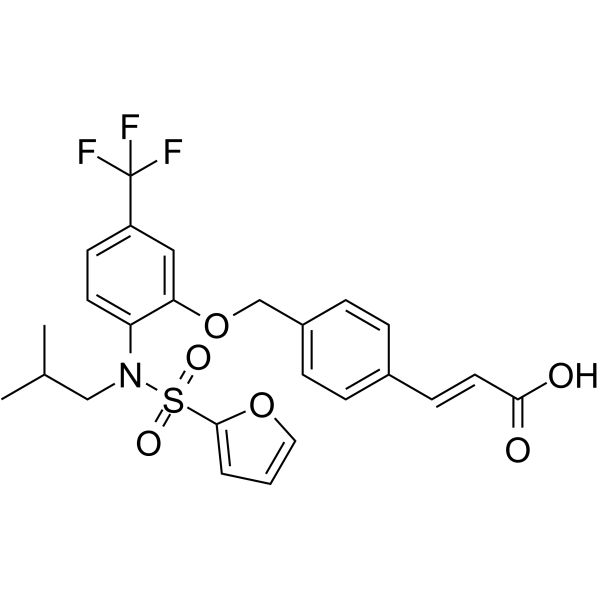
-
- HY-P1219
-
|
β-TRTX-Cj1α
|
Sodium Channel
|
Neurological Disease
|
|
Jingzhaotoxin-III is a potent and selective blocker of Nav1.5 channels, with an IC50 of 348 nM, and shows no effect on other sodium channel isoforms. Jingzhaotoxin-III can selectively inhibit the activation of cardiac sodium channel but not neuronal subtypes, and hopefully represents an important ligand for discriminating cardiac VGSC subtype .
|
-

-
- HY-125382
-
-
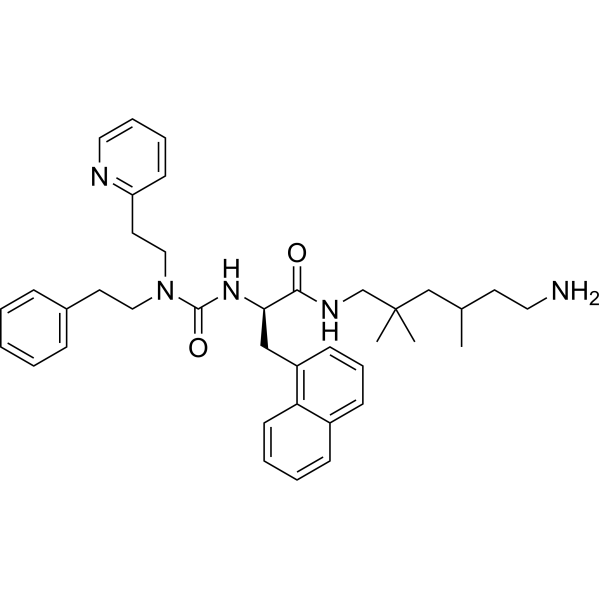
-
- HY-163317
-
|
|
Somatostatin Receptor
|
Cancer
|
|
MMC(TMZ)-TOC has high binding affinity and selectivity for somatostatin receptor subtype-2 (SSTR2). MMC(TMZ)-TOC targets delivery of TMZ to SSTR2-positive tumor cells .
|
-
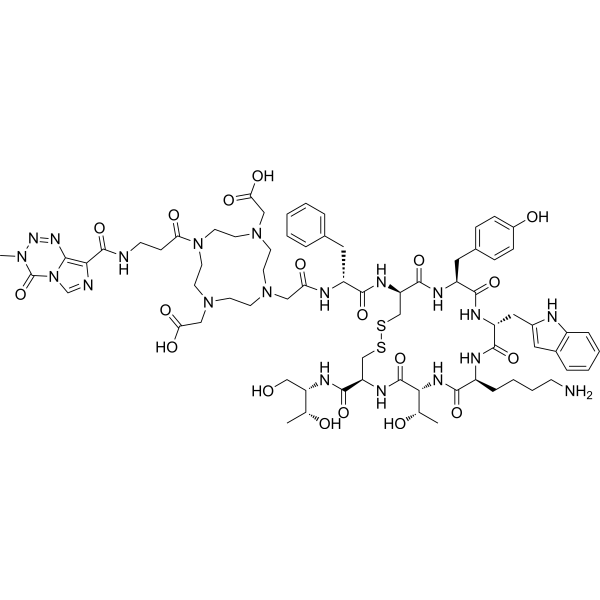
-
- HY-B0374
-
-
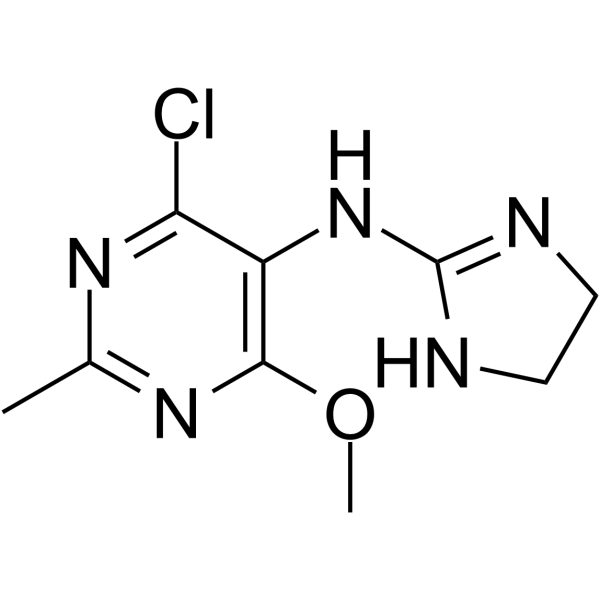
-
- HY-12811
-
|
|
Sodium Channel
|
Neurological Disease
|
|
PF-04856264 is a potent and selective Nav1.7 inhibitor, with IC50s of 28, 131, 19, and 42 nM for human, mouse, cynomolgus monkey and dog Nav1.7, respectively. PF-04856264 has low potency against the rat Nav1.7 channel. PF-04856264 shows analgesic effect .
|
-
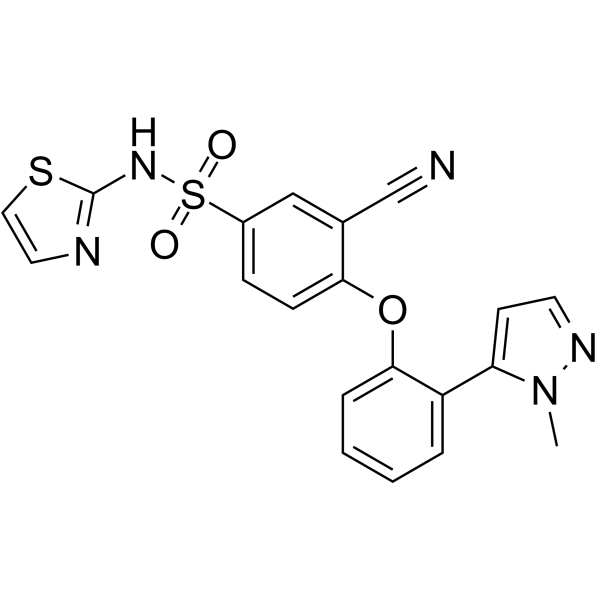
-
- HY-B0197
-
|
GR-85548A
|
5-HT Receptor
|
Neurological Disease
|
|
Naratriptan is a selective 5-HT1 receptor subtype agonist and is a triptan drug that is used for the treatment of migraine headaches.
|
-
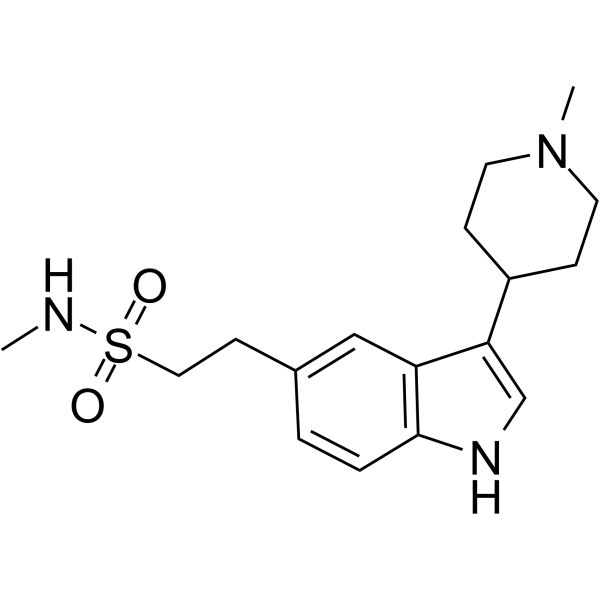
-
- HY-B0197A
-
|
GR-85548A hydrochloride
|
5-HT Receptor
|
Neurological Disease
|
|
Naratriptan hydrochloride is a selective 5-HT1 receptor subtype agonist and is a triptan drug that is used for the treatment of migraine headaches.
|
-
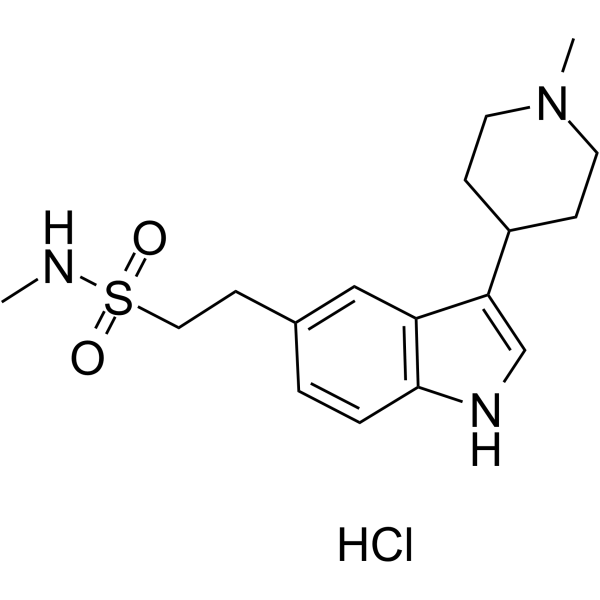
-
- HY-142908
-
|
|
Endogenous Metabolite
|
Cancer
|
|
Maximiscin, a fungal metabolite, induces DNA damage and shows selective cytotoxic activity against a subtype of triple-negative breast cancer.
|
-
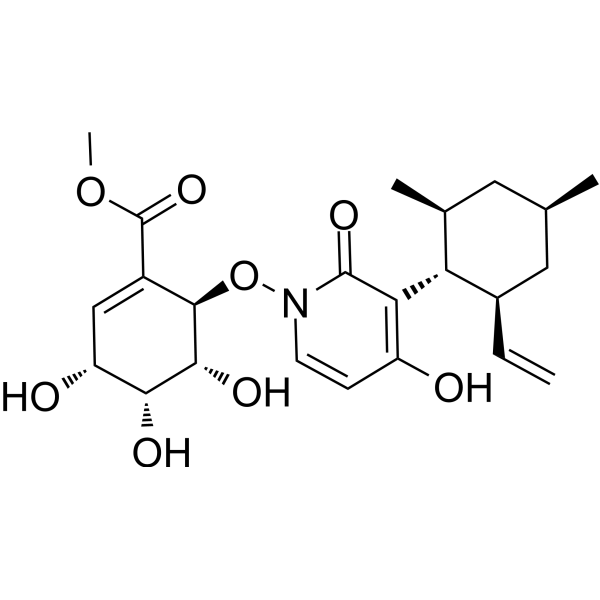
-
- HY-B0197AS
-
-
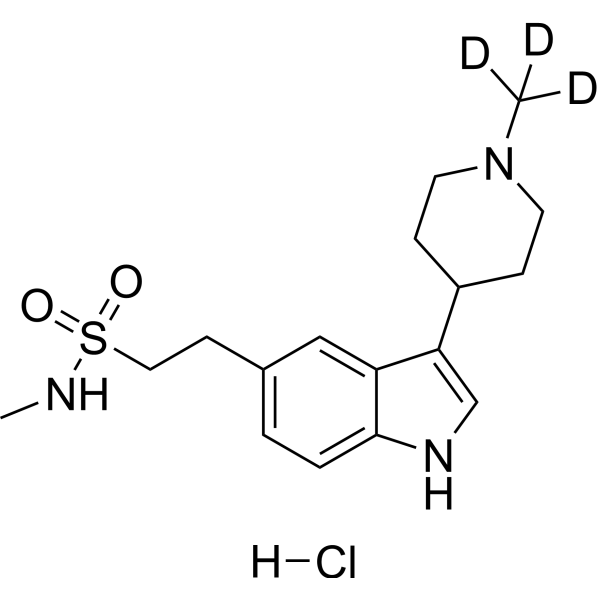
-
- HY-12439
-
ML380
1 Publications Verification
|
mAChR
|
Neurological Disease
|
|
ML380 is a potent, subtype-selective, and brain-penetrant positive allosteric modulator (PAM) of M5 mAChR, with EC50s of 190 and 610 nM for human and rat M5, respectively. ML380 exhibits moderate selectivity versus the M1 and M3 mAChR subtypes. ML380 could increase the affinity of ACh for the M5 mAChR .
|
-
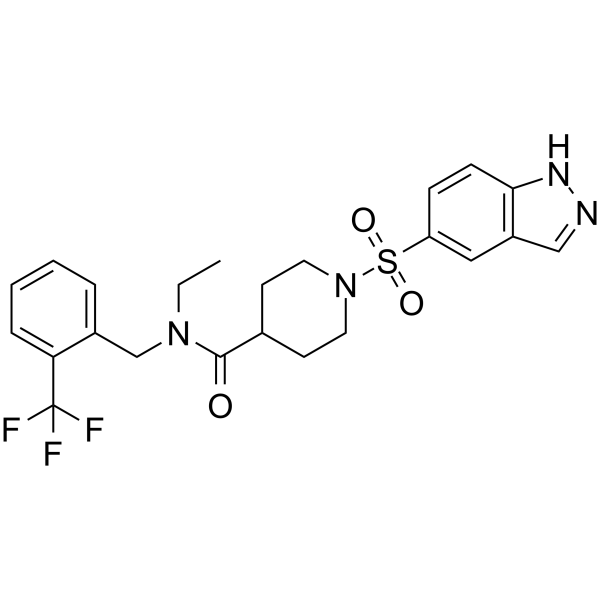
-
- HY-162117
-
|
|
mGluR
|
Neurological Disease
|
|
LBG30300 is a subtype-selective mGlu2 receptor agonist EC50 0.6 nM. LBG30300 is blood-brain barrier permeable .
|
-
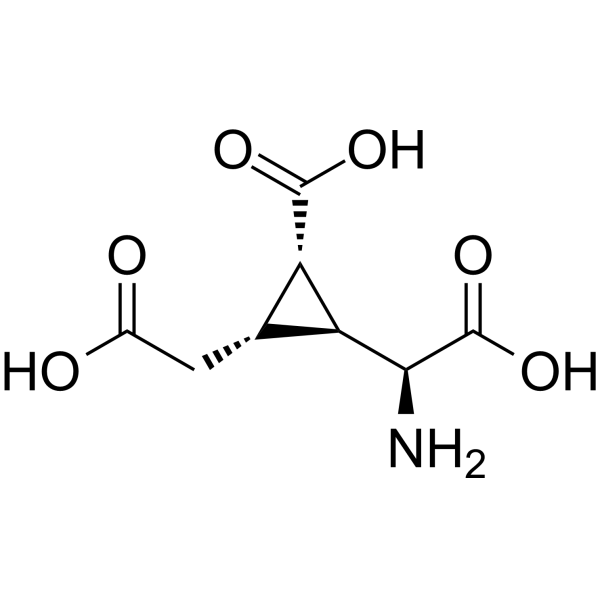
-
- HY-101869
-
|
MRK-409
|
GABA Receptor
|
Neurological Disease
|
|
MK0343 (MRK-409) is an orally bioavailable GABAA receptor subtype-selective partial agonist. MK0343 is a non-sedating anxiolytic .
|
-
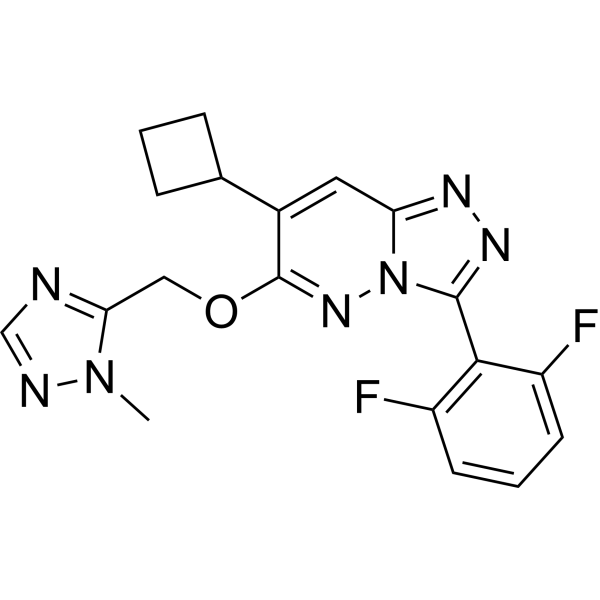
-
- HY-157889
-
|
|
HDAC
|
Cancer
|
|
ZINC000028464438 is a selective HDAC11 inhibitor with an IC50 of 3.5 µM. ZINC000028464438 shows almost no inhibition for other HDAC subtypes .
|
-
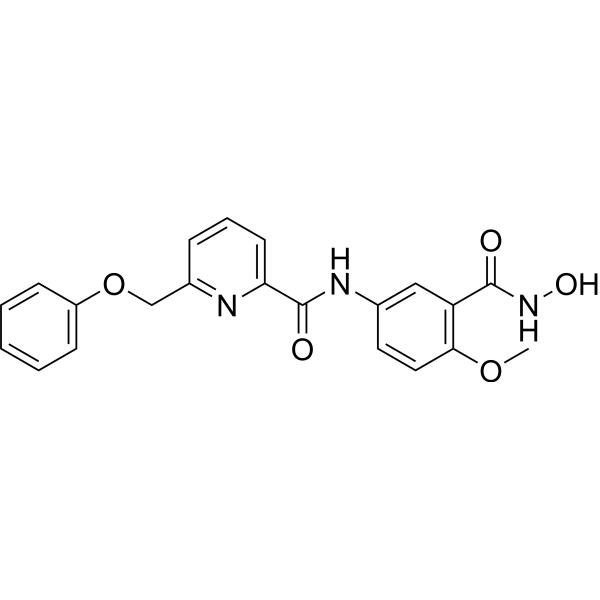
-
- HY-101640
-
-
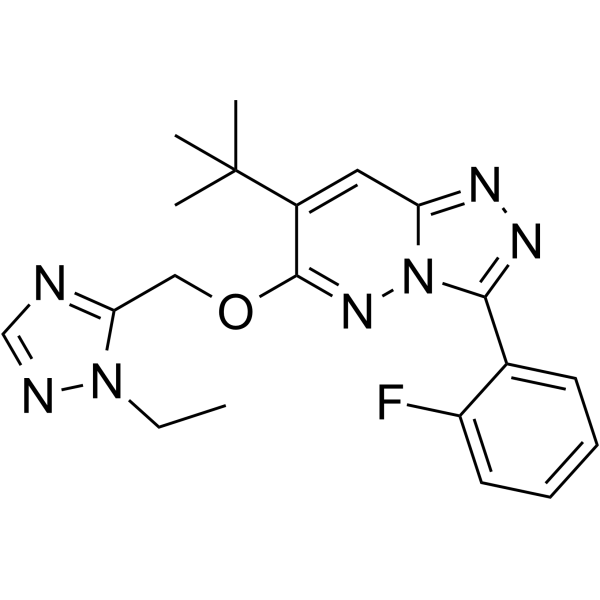
-
- HY-108498
-
|
|
Somatostatin Receptor
|
Endocrinology
|
|
L-817818 is a potent and subtype-selective agonist of the somatostatin receptor. L-817818 provides a direct approach to defining somatostatin receptor physiological functions
|
-
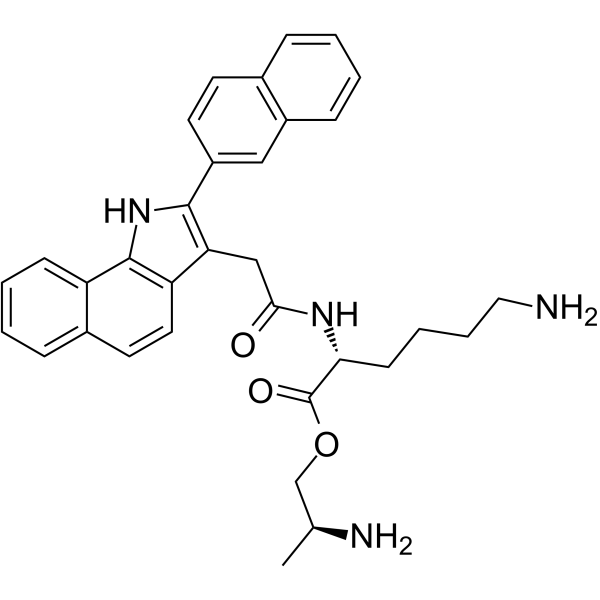
-
- HY-107713
-
|
|
iGluR
|
Neurological Disease
|
|
PPDA is a subtype-selective NMDA receptor antagonist that preferentially binds to NR2C/NR2D containing receptors .
|
-
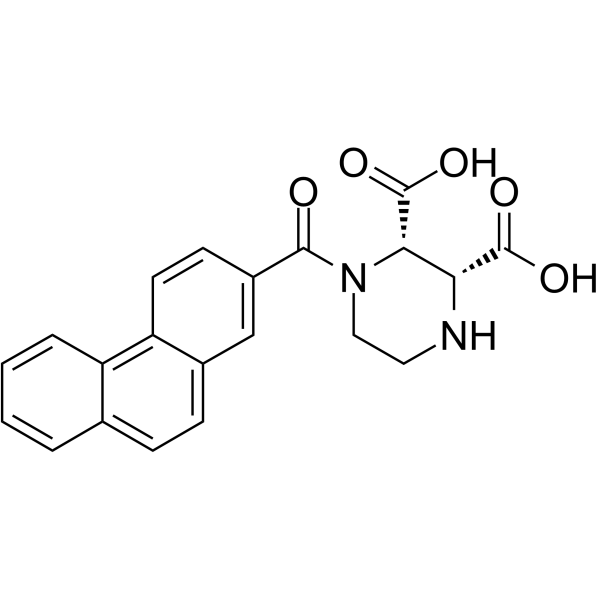
-
- HY-B0197S
-
|
GR-85548A-d3-1
|
5-HT Receptor
|
Neurological Disease
|
|
Naratriptan-d3 is the deuterium labeled Naratriptan[1]. Naratriptan is a selective 5-HT1 receptor subtype agonist[2].
|
-
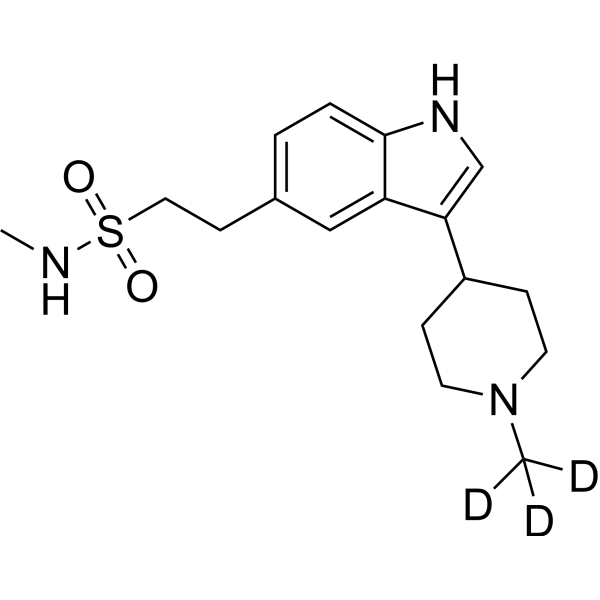
-
- HY-111271A
-
-
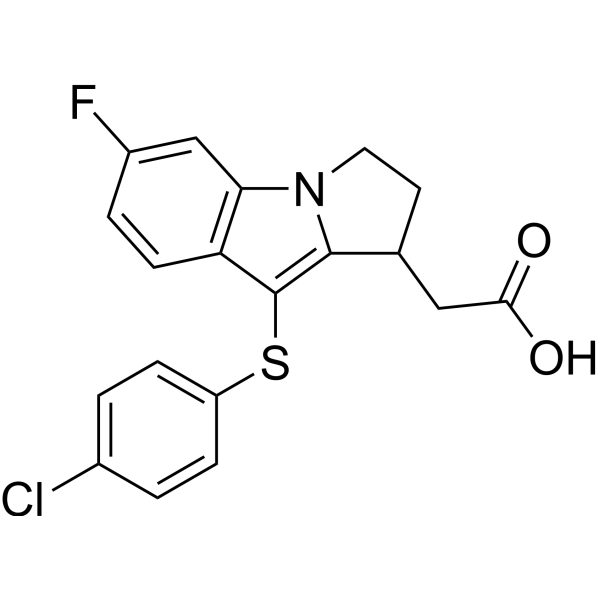
-
- HY-117196
-
-
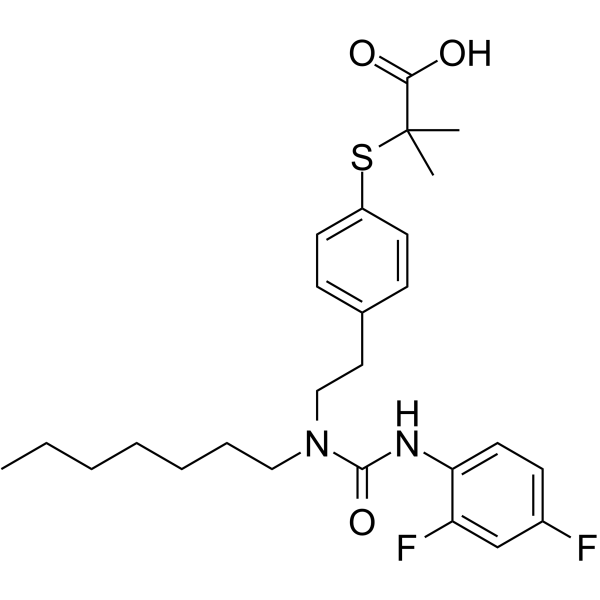
-
- HY-147664
-
|
|
Carbonic Anhydrase
|
Cancer
|
|
These compounds show selective inhibition on tumor related subtypes HCA IX and XII, and are also considered as the leading molecules for the development of future cancer therapeutic drugs based on new mechanisms of action.
|
-
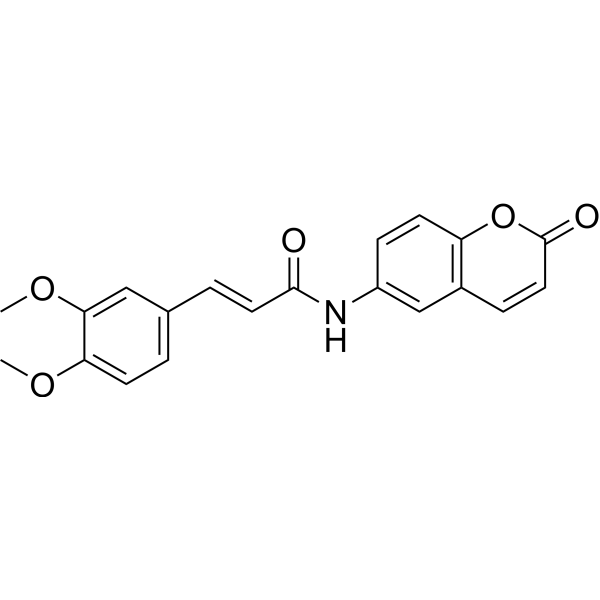
-
- HY-15393
-
|
|
mGluR
|
Neurological Disease
|
|
VU 0357121 is a positive and highly selective mGlu5R allosteric modulator (PAM) with an EC50 of 33 nM. VU 0357121 is inactive or very weakly antagonizing at other mGlu receptor subtypes .
|
-
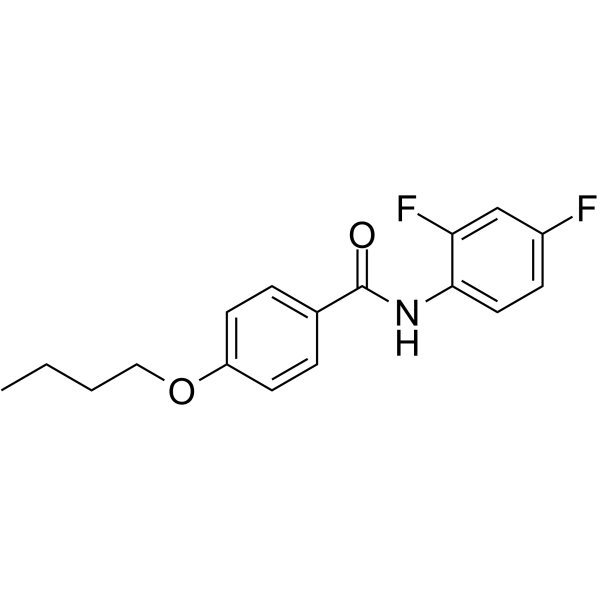
-
- HY-N0584A
-
|
6-Hydroxyhyoscyamine hydrobromide
|
mAChR
|
Inflammation/Immunology
|
|
Anisodamine hydrobromide (6-Hydroxyhyoscyamine hydrobromide), a belladonna alkaloid, is a non-subtype-selective muscarinic and a nicotinic cholinoceptor antagonist. Anisodamine hydrobromide shows antioxidant, anti-inflammatory properties .
|
-
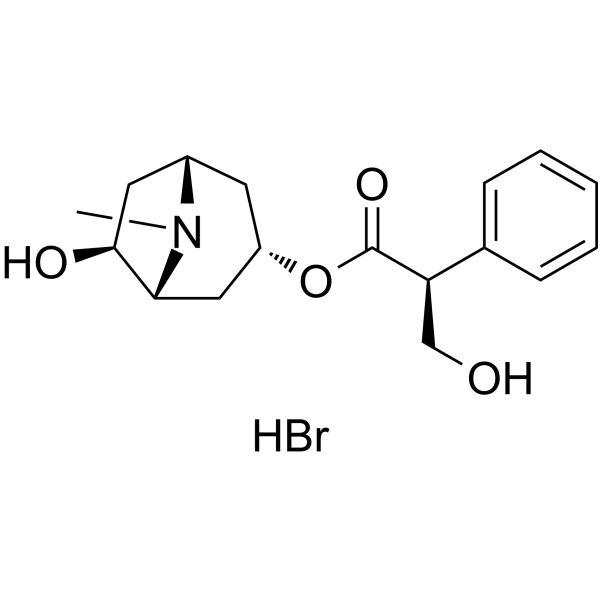
-
- HY-150727
-
|
|
Sirtuin
|
Cancer
|
|
SIRT5 inhibitor 4 (compound 11) is a potent, selective SIRT5 inhibitor with IC50 values of 26.4 and >400μM for SIRT5 and other SIRT subtype, respectively .
|
-
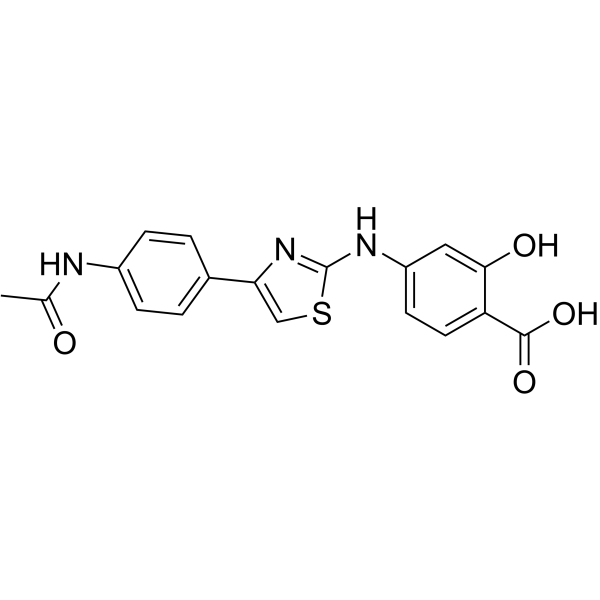
-
- HY-15072
-
|
YM 872
|
iGluR
|
Metabolic Disease
|
|
Zonampanel (YM 872) is a selective antagonist of the glutamate receptor subtype, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor.
|
-
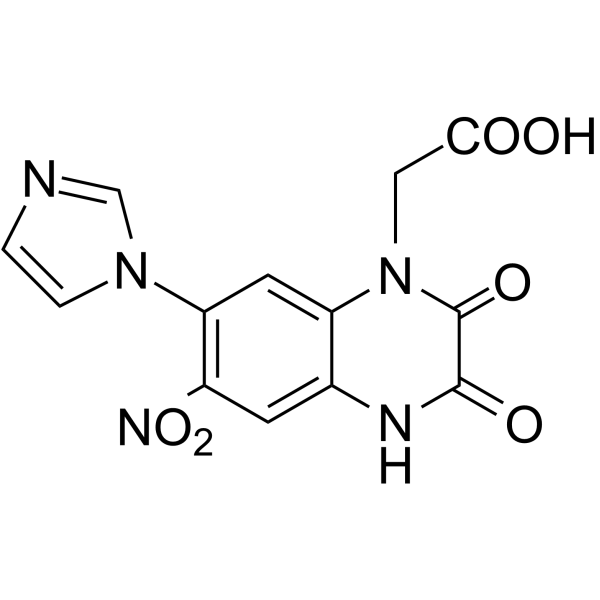
-
- HY-128770
-
|
|
Dopamine Receptor
|
Neurological Disease
|
|
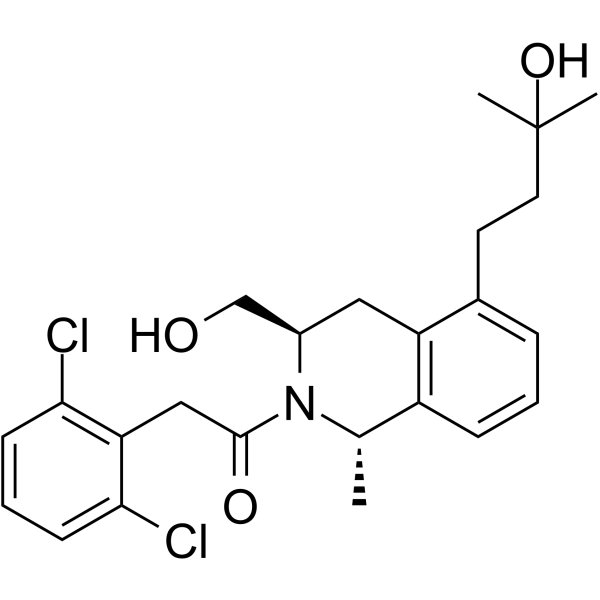
LY3154207 is a potent, subtype selective, and orally available human dopamine D1 receptor
positive allosteric modulator (PAM) with minimal allosteric agonist activity (EC50=3 nM) .
|
-
-
- HY-D0976
-
|
|
P2X Receptor
HIV
|
Infection
|
|
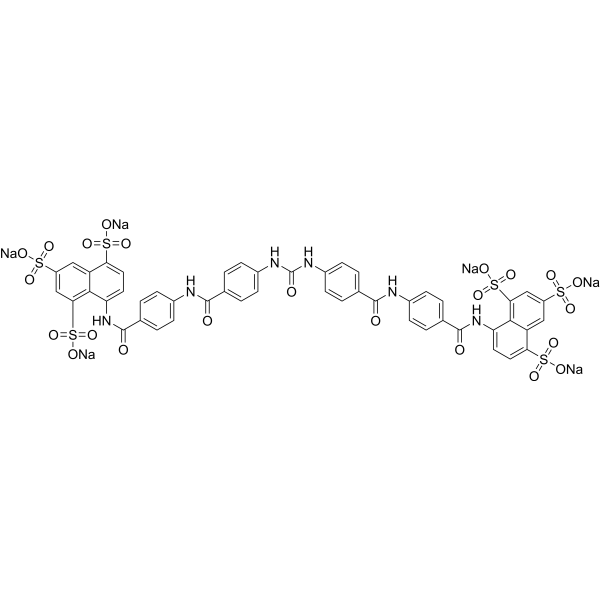
NF279 is a potent selective and reversible P2X1 receptor antagonist, with an IC50 of 19 nM. NF279 displays good selectivity over P2X2, P2X3 (IC50=1.62 μM), P2X4 (IC50>300 μM). NF279 is a dual HIV-1 coreceptor inhibitor that interferes with the functional engagement of CCR5 and CXCR4 by Env .
|
-
-
- HY-14342
-
MK-5046
1 Publications Verification
|
Bombesin Receptor
|
Metabolic Disease
|
|
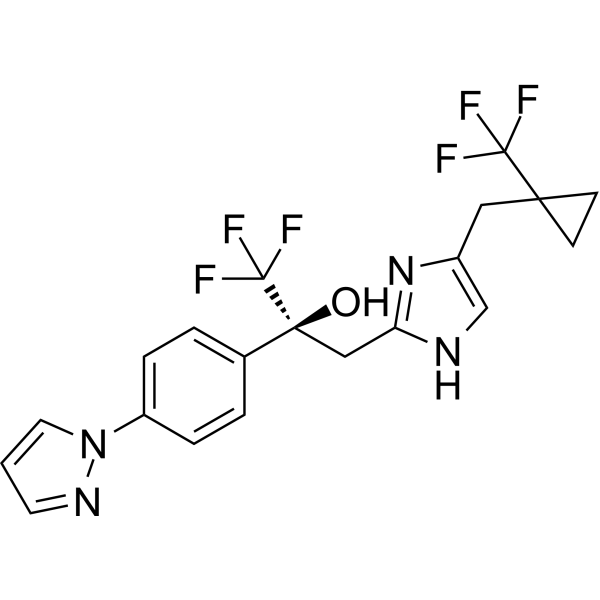
MK-5046 is a potent, selective and orally active Bombesin receptor subtype-3 (BRS-3) allosteric agonist with an IC50 and an EC50 value of 27 and 25 nM for hBRS-3, respectively. MK-5046 inhibits food intake and reduces body weight of diet-induced obese (DIO) mouse models. MK-5046 can be used for the research of obesity .
|
-
-
- HY-116046
-
|
|
Melatonin Receptor
|
Others
|
|
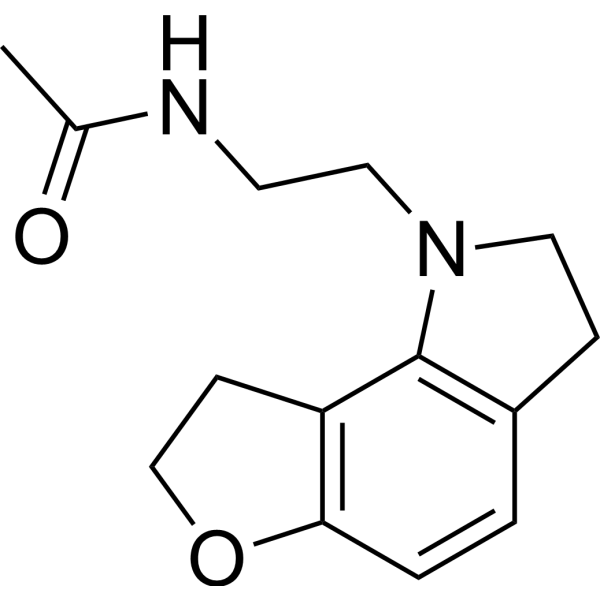
GR 196429 is a melatonin receptor agonist with some selectivity for the MT1 subtype. GR 196429 produces both sleep-promoting effects and alterations of circadian rhythm, as well as stimulating melatonin release in mice .
|
-
-
- HY-B1154
-
-
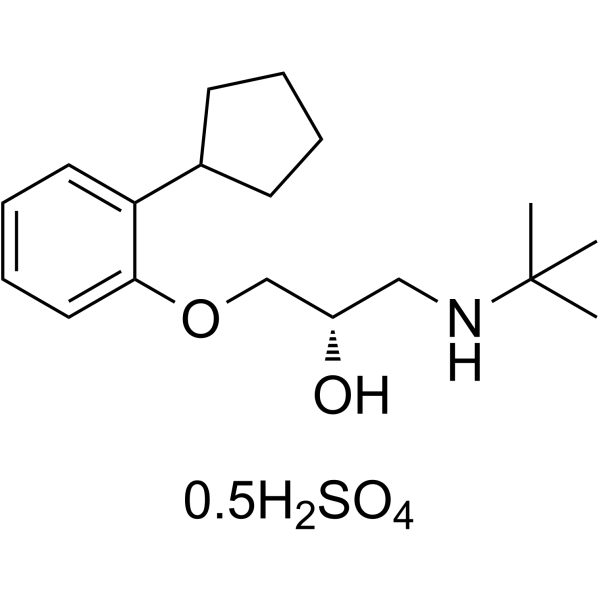
-
- HY-108710
-
|
|
mGluR
|
Neurological Disease
|
|
VU0650786 is a potent and selective CNS penetrant negative allosteric modulator of metabotropic glutamate receptor subtype 3 (mGlu3 NAM), with an IC50 of 392 nM. VU0650786 has antidepressant and anxiolytic activity in rodents .
|
-
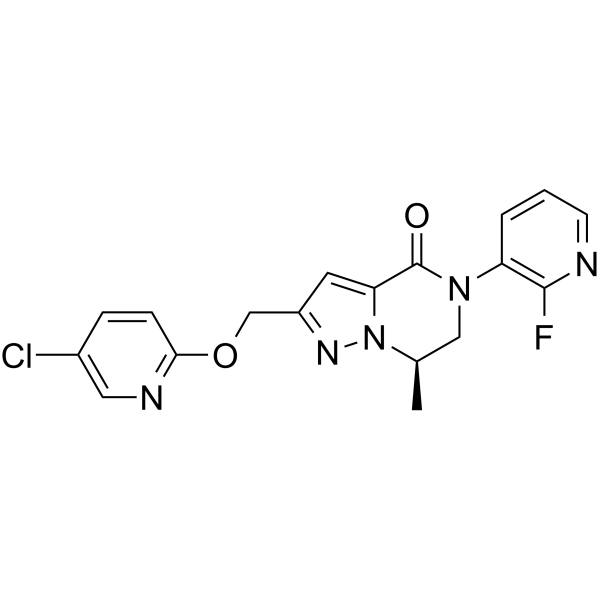
-
- HY-114191B
-
|
|
Somatostatin Receptor
|
Metabolic Disease
Endocrinology
|
|
SSTR5 antagonist 2 hydrochloride is a highly potent, oral active and selective somatostatin (receptor) subtype 5 (SSTR5) antagonist and has potential for the research of type 2 diabetes mellitus (T2DM) .
|
-
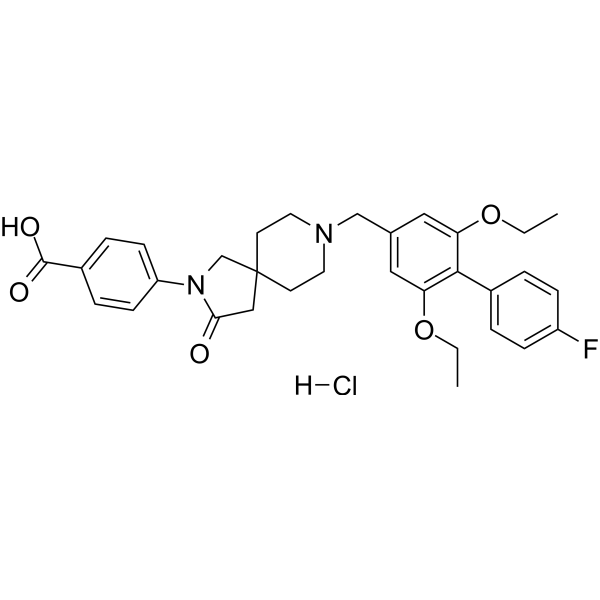
-
- HY-110131
-
|
|
nAChR
|
Neurological Disease
|
|
A 85380 hydrochloride is a novel, high affinity neuronal nicotinic acetylcholine receptor (nAChR) agonist. A 85380 hydrochloride exhibits selectivity for the α4β2 nAChR subtypes. A 85380 hydrochloride has a broad-spectrum analgesic profile .
|
-
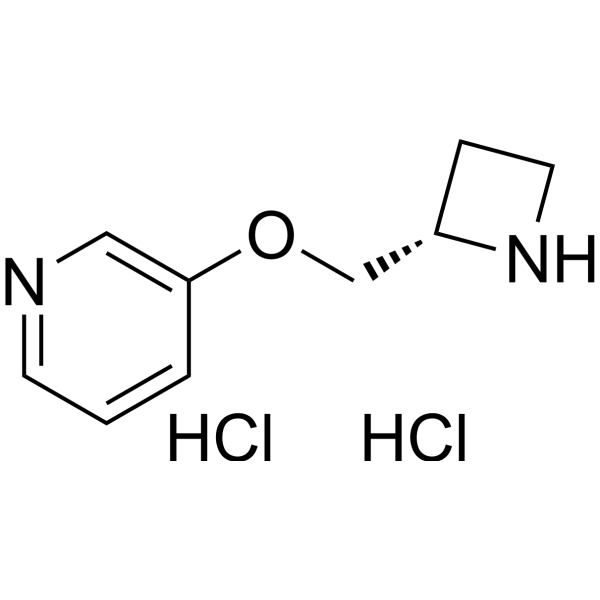
-
- HY-14565
-
|
ABT-089
|
nAChR
|
Neurological Disease
|
|
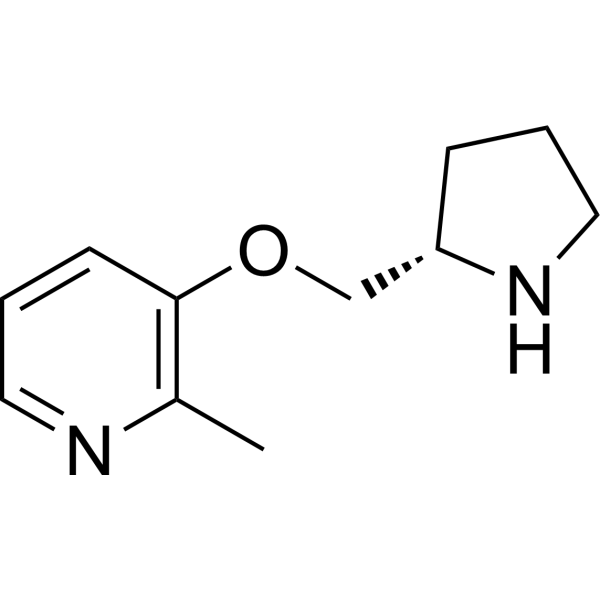
Pozanicline (ABT-089) selectively activate neuronal nicotinic acetylcholine receptor (nAChR) subtypes, is a novel cholinergic agent that is a partial agonist at α4β2* nAChRs (Ki=16 nM) and shows high selectivity for α6β2* and α4α5β2 nAChR subtypes, the binding affinity (Ki, rat) for Pozanicline to [ 3H] cytisine sites is 16.7 nM.
Pozanicline reverses nicotine withdrawal-induced cognitive deficits, may be an effective component of novel therapeutic strategies for nicotine addiction .
|
-
-
- HY-102037
-
-
-
- HY-W580721A
-
|
|
Histamine Receptor
|
Others
|
|
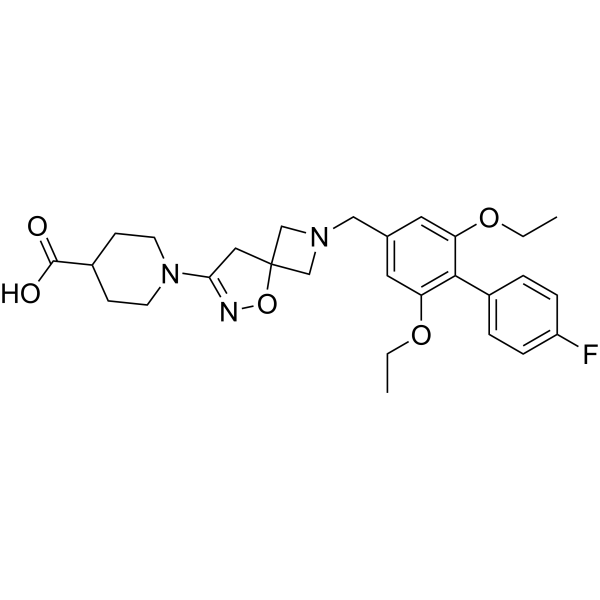
4-Methylhistamine (hydrochloride) is a potent, high affinity H4 receptor agonist Ki of 7 nM. 4-Methylhistamine (hydrochloride) displays more than 100-fold selectivity over other human histamine receptor subtypes .
|
-
-
- HY-13225
-
|
RJR-2403 oxalate; (E)-Metanicotine oxalate
|
nAChR
|
Neurological Disease
|
|
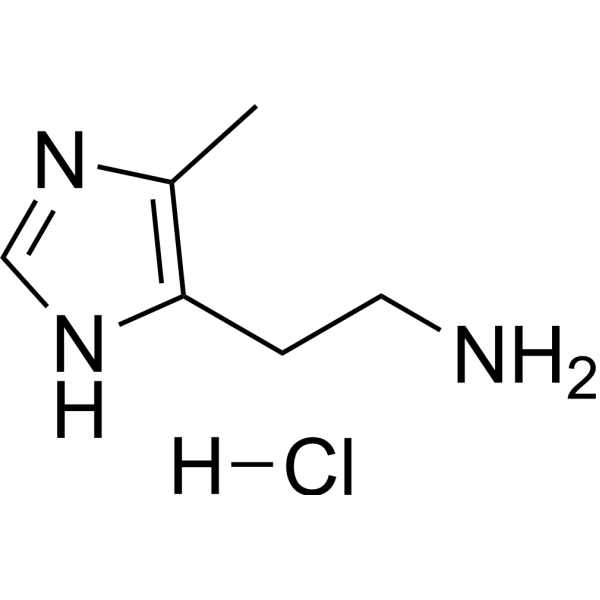
Rivanicline oxalate (RJR-2403 oxalate; (E)-Metanicotine oxalate) is a neuronal nicotinic receptor agonist, showing high selectivity for the α4β2 subtype (Ki=26 nM); > 1,000 fold selectivity than α7 receptors(Ki= 3.6 μM).
|
-
-
- HY-13225B
-
|
RJR-2403 hemioxalate; (E)-Metanicotine hemioxalate
|
nAChR
|
Neurological Disease
|
|
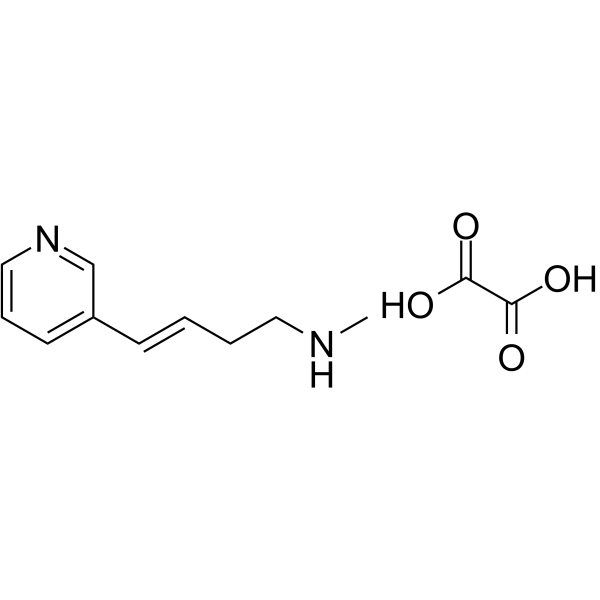
Rivanicline hemioxalate (RJR-2403 hemioxalate; (E)-Metanicotine hemioxalate) is a neuronal nicotinic receptor agonist, showing high selectivity for the α4β2 subtype (Ki=26 nM); > 1,000 fold selectivity than α7 receptors(Ki= 3.6 μM).
|
-
-
- HY-108592
-
|
|
Potassium Channel
|
Neurological Disease
|
|
UCL 2077 is a selective slow-afterhyperpolarization (sAHP) channel blocker (IC50 = 500 nM in hippocampal neurons in culture), having minimal effects on Ca2+ channels, action potentials, input resistance and the medium after hyperpolarization . UCL 2077 is also a subtype-selective blocker of the epilepsy associated KCNQ channels .
|
-
-
- HY-117960
-
|
LEO-32731
|
Phosphodiesterase (PDE)
|
Inflammation/Immunology
|
|
Orismilast (LEO-32731) is an orally active and selective PDE4 inhibitor used for the research of inflammatory diseases. Orismilast demonstrates potent inhibition of PDE4B and PDE4D subtype splice variants .
|
-
- HY-114191
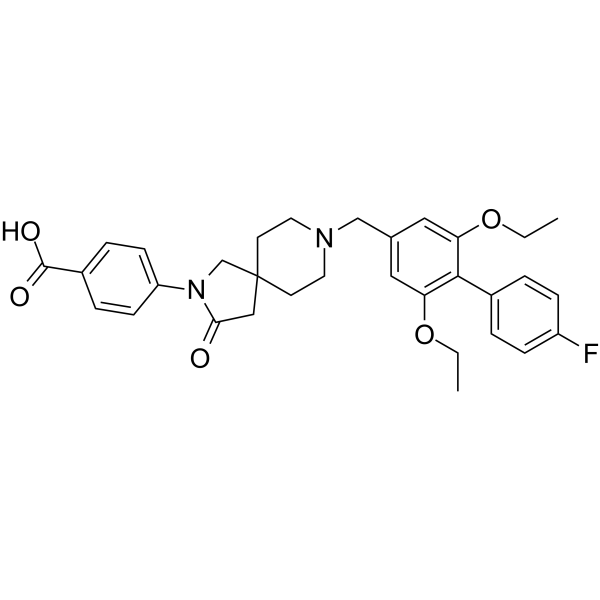
-
|
|
Somatostatin Receptor
|
Metabolic Disease
Endocrinology
|
|
SSTR5 antagonist 2 (compound 10) is a highly potent, oral active and selective somatostatin (receptor) subtype 5 (SSTR5) antagonist and has potential for the research of treat type 2 diabetes mellitus (T2DM) .
|
-
- HY-B0374S
-
|
|
Imidazoline Receptor
|
Neurological Disease
|
|
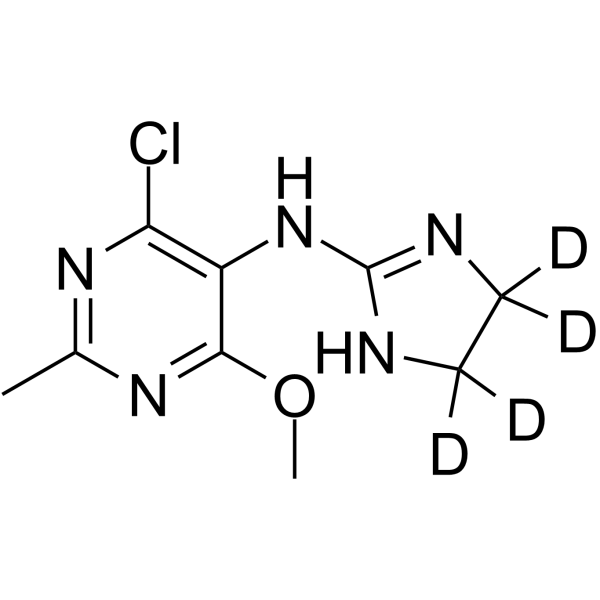
Moxonidine-d4 (BDF5895-d4) is the deuterium labeled Moxonidine. Moxonidine(BDF5895) is a selective agonist at the imidazoline receptor subtype 1, used as antihypertensive agent[1][2].
|
-
- HY-114191A
-
|
|
Somatostatin Receptor
|
Metabolic Disease
Endocrinology
|
|
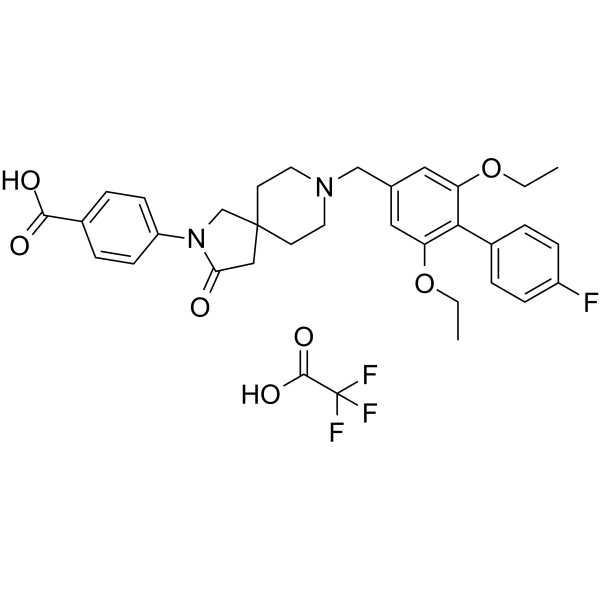
SSTR5 Antagonist 1 (compound 10) is a highly potent, oral active and selective somatostatin (receptor) subtype 5 (SSTR5) antagonist and has potential for the research of treat type 2 diabetes mellitus (T2DM) .
|
-
- HY-103183
-
-
- HY-15688
-
|
8-Hydroxy-DPAT hydrobromide
|
5-HT Receptor
|
Neurological Disease
|
|
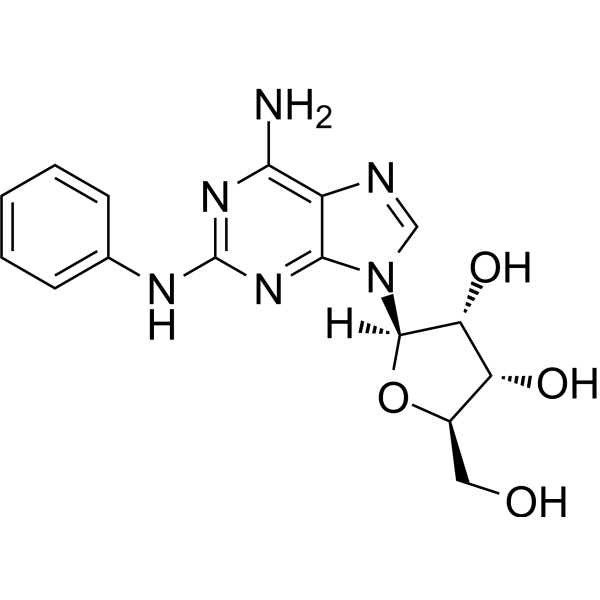
8-OH-DPAT (8-Hydroxy-DPAT) hydrobromide is a potent and selective 5-HT1A agonist with a pIC50 of 8.19. 8-OH-DPAT hydrobromide has selectivity of almost 1000 fold for a subtype of the 5-HT1 binding site .
|
-
- HY-N0584
-
|
6-Hydroxyhyoscyamine
|
|
|
|
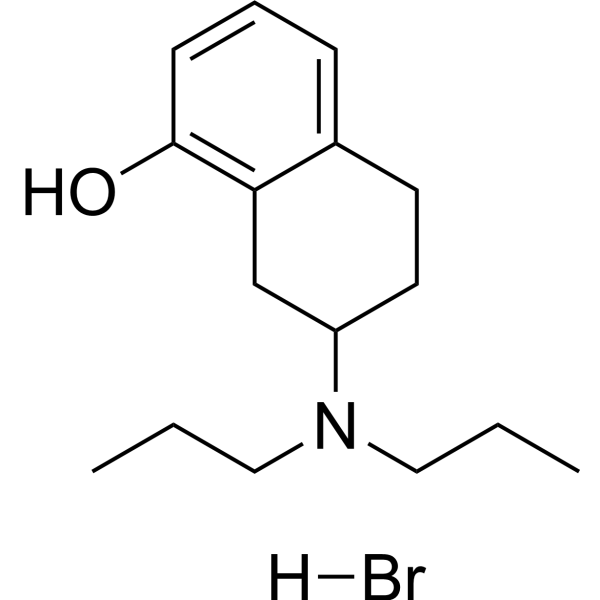
Anisodamine (6-Hydroxyhyoscyamine), a belladonna alkaloid, is a non-subtype-selective muscarinic, and also a nicotinic cholinoceptor antagonist. Anisodamine employs in traditional Chinese medicine for many ailments, mainly to improve the microcirculation in states of shock, and also in organophosphate poisoning .
|
-
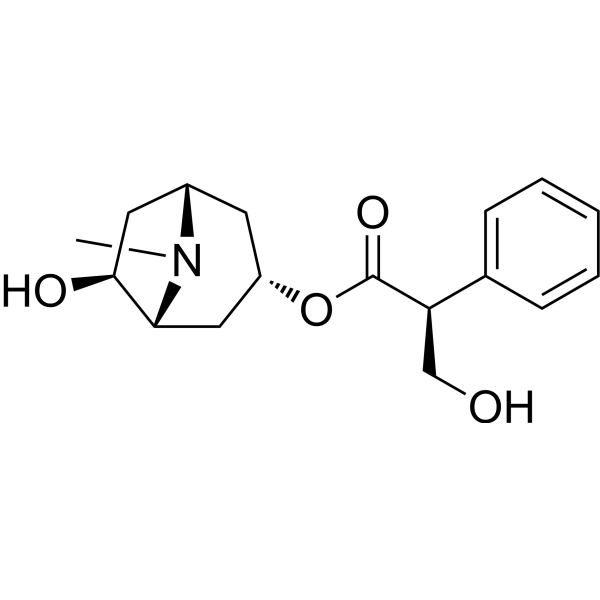
- HY-P5158
-
|
|
Adrenergic Receptor
|
Others
|
|
Conopeptide rho-TIA is a peptide derived from the venom contained in the predatory sea snail Conus tulipa, has highly selective and noncompetitive inhibitor at human α1B-Adrenergic Receptor. Conopeptide rho-TIA acts a competitive inhibitor at human α1A-Adrenergic Receptor and α1D-Adrenergic Receptor. Conopeptide rho-TIA binds to each subtype and may provide useful information for the development of novel α1-Adrenergic Receptor subtype-selective drugs .
|
-
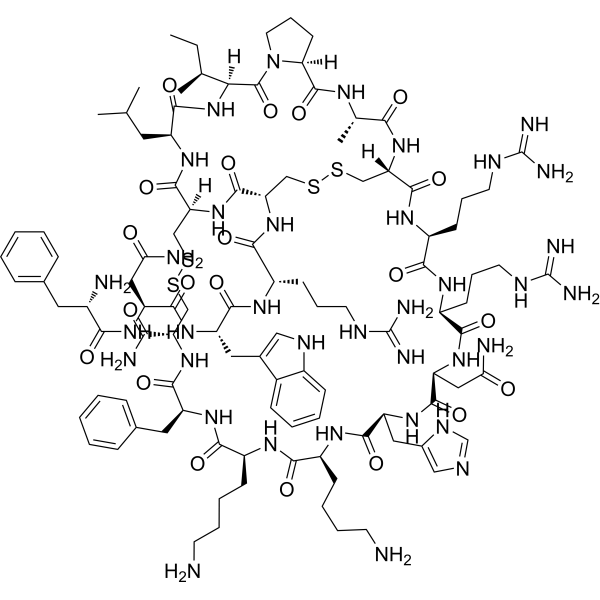
- HY-107457
-
|
|
mGluR
|
Neurological Disease
|
|
AZD-8529 is a potent, highly selective and orally bioavailable positive allosteric modulator of mGluR2, with an EC50 of 285 nM, and shows no positive allosteric modulator responses at 20-25 M on the mGluR1, 3, 4, 5, 6, 7, and 8 subtypes.
|
-
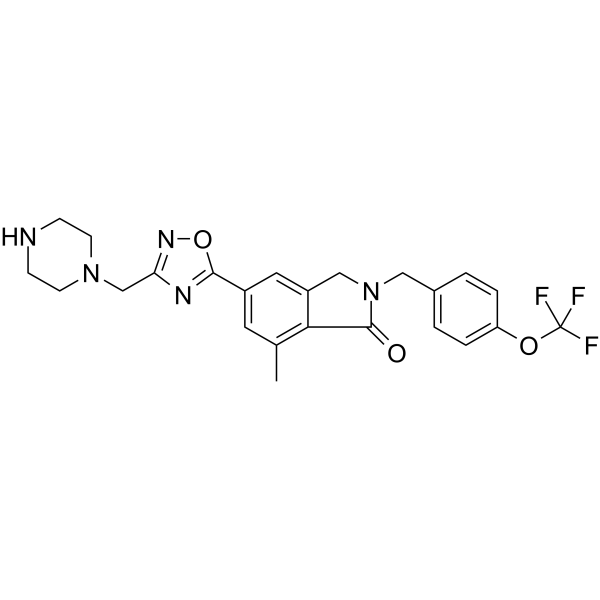
- HY-136459
-
|
|
iGluR
|
Neurological Disease
|
|
NMDA receptor antagonist 2 is a potent and orally active NR2B subtype-selective NMDA antagonist with an IC50 and a Ki of 1.0 nM and 0.88 nM, respectively. NMDA receptor antagonist 2 is used for the study of neuropathic pain and Parkinson’s disease .
|
-
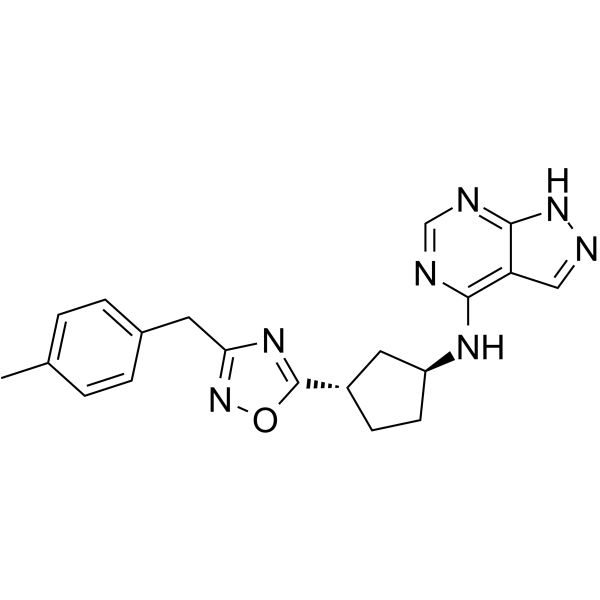
- HY-102094
-
|
|
mGluR
|
Neurological Disease
|
|
(E/Z)-SIB-1893 is a racemic compound of (E)-SIB-1893 and (Z)-SIB-1893 isomers. (E)-SIB-1893 is a selective non-competitive metabotropic glutamate subtype 5 receptor (mGluR5) antagonist .
|
-
- HY-115766
-
|
|
nAChR
|
Neurological Disease
|
|
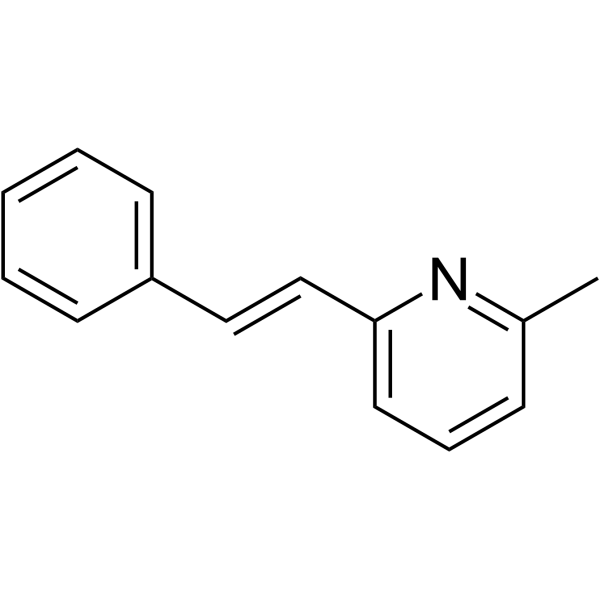
Anabaseine is a non-selective nicotinic agonist. Anabaseine stimulates all AChRs, preferentially stimulates skeletal muscle and brain α7 subtypes . Anabaseine is also a weak partial agonist at α4β2 nAChRs .
|
-
- HY-P5172
-
|
|
Sodium Channel
|
Neurological Disease
|
|
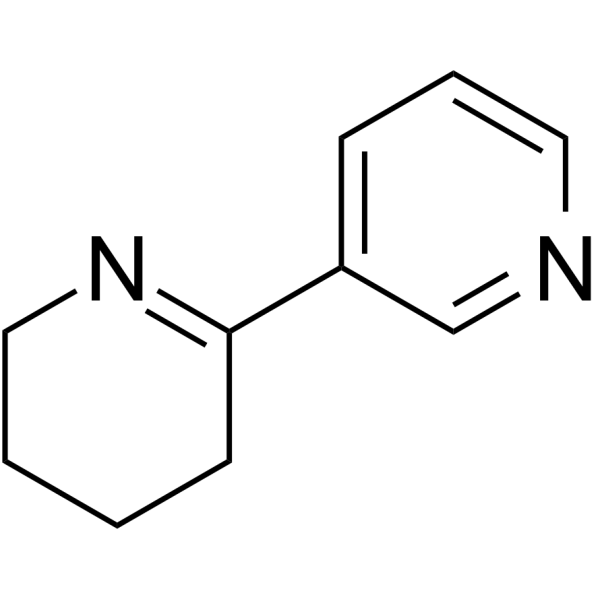
MitTx-alpha is a subunit of MitTx. MitTx is a potent, persistent, and selective agonist for acid-sensing ion channels (ASICs). MitTx is highly selective for the ASIC1 subtype at neutral pH; under more acidic conditions (pH<6.5), MitTx massively potentiates (>100-fold) proton-evoked activation of ASIC2a channels .
|
-

- HY-17015
-
|
RWJ 270201 trihydrate; BCX 1812 trihydrate
|
Influenza Virus
|
Infection
|
|
Peramivir trihydrate (RWJ-270201 trihydrate;BCX-1812 trihydrate) is a highly potent, selective and orally active influenza virus neuraminidase (NA) inhibitor, with IC50 values ranging from 0.9 to 4.3 nM for nine NA subtypes .
|
-
- HY-107457A
-
|
|
mGluR
|
Neurological Disease
|
|
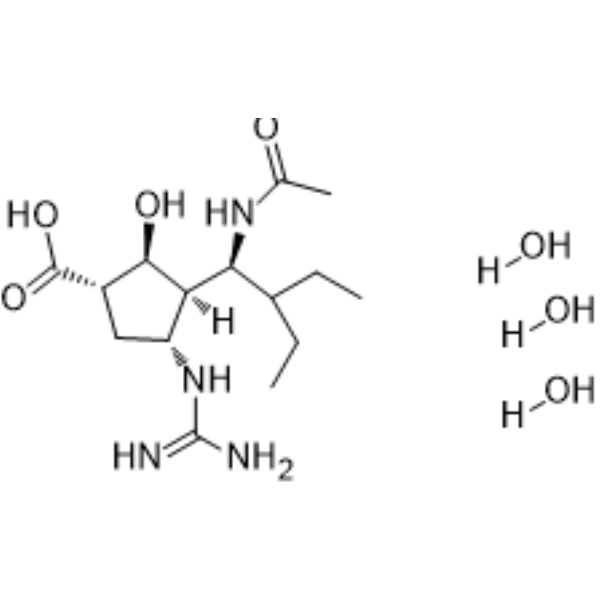
AZD-8529 mesylate is a potent, highly selective and orally bioavailable positive allosteric modulator of mGluR2, with an EC50 of 285 nM, and shows no positive allosteric modulator responses at 20-25 M on the mGluR1, 3, 4, 5, 6, 7, and 8 subtypes .
|
-
- HY-14569
-
CDPPB
1 Publications Verification
|
mGluR
|
Neurological Disease
|
|
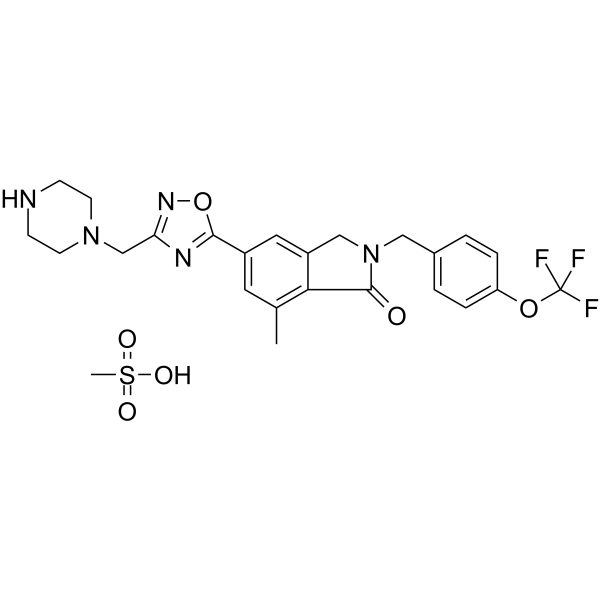
CDPPB is a potent, selective and brain penetrant positive allosteric modulator of the metabotropic glutamate receptor subtype 5 (mGluR5), with an EC50 of 27 nM in Chinese hamster ovary cells expressing human mGluR5. CDPPB may provide an approach for development of antipsychotic agents .
|
-
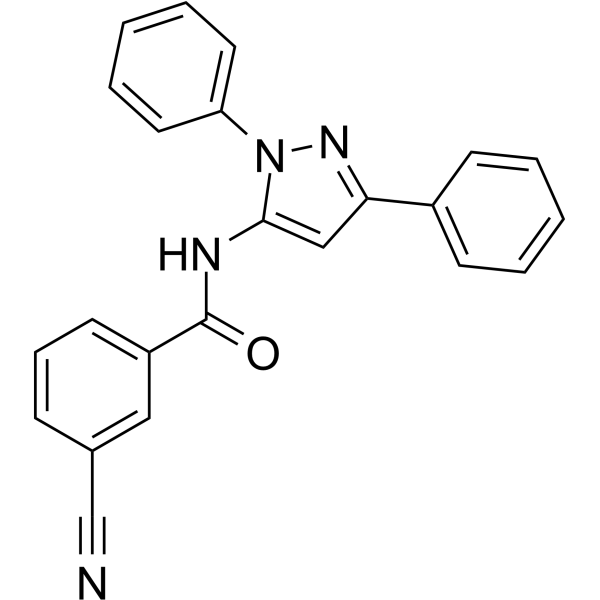
- HY-103235
-
|
|
iGluR
|
Neurological Disease
|
|
NPEC- caged-(S)-AMPA, a caged neurotransmitter analog, is a NPEC photoprotecting group caged the (S)-AMPA (HY-100815A) to make caged ligands specific for glutamate receptor sub-types. NPEC- caged-(S)-AMPA selectively activates AMPA receptor .
|
-
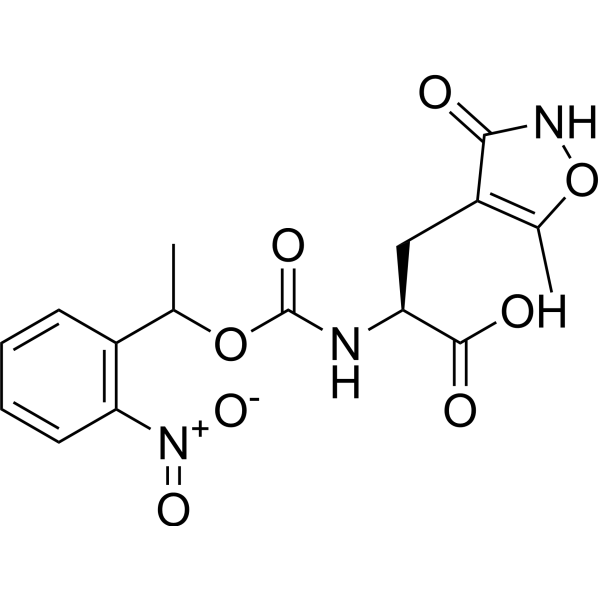
- HY-14277A
-
|
R 50547 hydrochloride
|
Histamine Receptor
Neurotensin Receptor
Integrin
|
Inflammation/Immunology
|
|
Levocabastine (R 50547) hydrochloride is a potent and selective histamine H1-receptor antagonist. Levocabastine hydrochloride is also a selective, high affinity neurotensin receptor subtype 2 (NTR2) antagonist, with a Ki of 17 nM for mNTR2. Levocabastine hydrochloride can act as a VLA-4 antagonist, interferes with conjunctival eosinophil infiltration in allergic conjunctivitis (AC) .
|
-
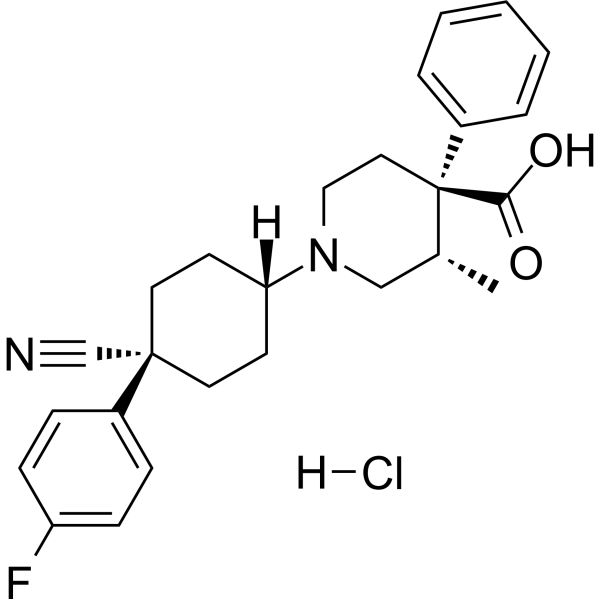
- HY-P1221
-
|
|
Sodium Channel
|
Neurological Disease
|
|
ProTx II is a selective blocker of Nav1.7 sodium channels with an IC50 of 0.3 nM, and is at least 100-fold selective for Nav1.7 over other sodium channel subtypes. ProTx-II inhibits sodium channels by decreasing channel conductance and shifting activation to more positive potentials and blocks action potential propagation in nociceptors .
|
-
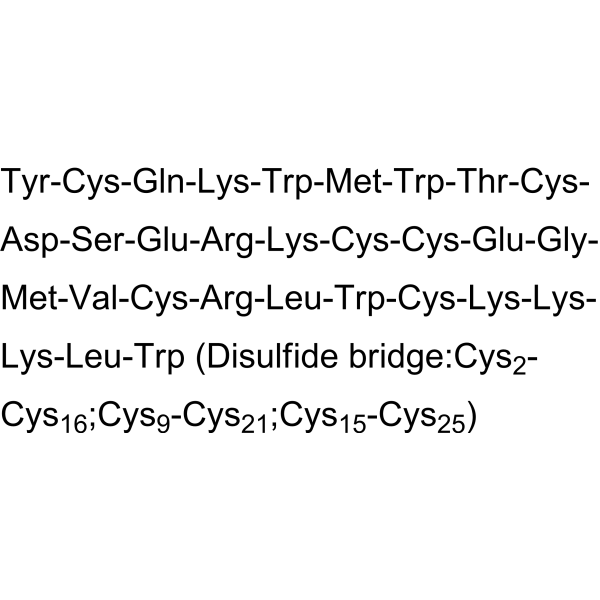
- HY-110092
-
|
|
P2Y Receptor
|
Metabolic Disease
|
|
PSB-1114 tetrasodium is a potent, enzymatically stable, and subtype-selective P2Y2 receptor agonist with an EC50 of 134 nM. PSB-1114 tetrasodium displays >50-fold selectivity versus the P2Y4 (EC50 of 9.3 μM) and P2Y6 (EC50 of 7.0 μM) receptors .
|
-
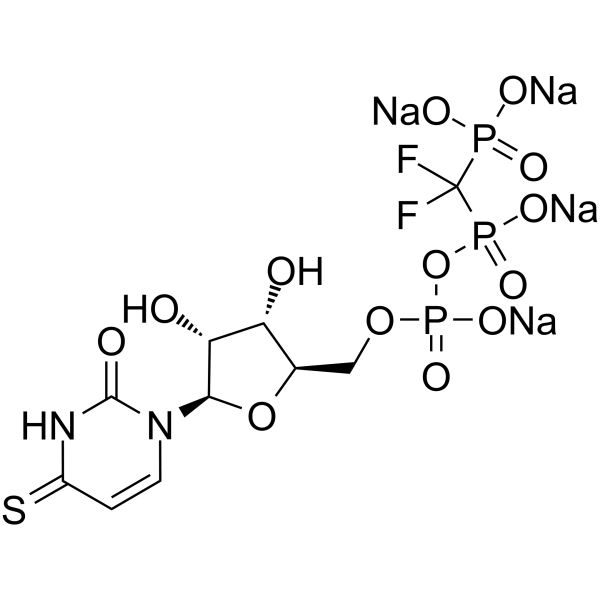
- HY-14277
-
|
R 50547
|
Histamine Receptor
Neurotensin Receptor
Integrin
|
Inflammation/Immunology
|
|
Levocabastine (R 50547) is a potent and selective histamine H1-receptor antagonist. Levocabastine hydrochloride is also a selective, high affinity neurotensin receptor subtype 2 (NTR2) antagonist, with a Ki of 17 nM for mNTR2. Levocabastine can act as a VLA-4 antagonist, interferes with conjunctival eosinophil infiltration in allergic conjunctivitis (AC) .
|
-
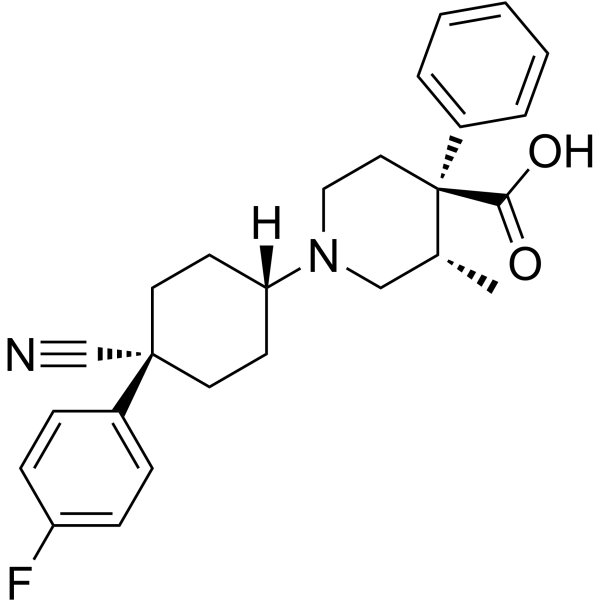
- HY-110092A
-
|
|
P2Y Receptor
|
Cancer
|
|
PSB-1114 triethylamine is a potent, enzymatically stable, and subtype-selective P2Y2 receptor agonist with an EC50 of 134 nM. PSB-1114 triethylamine displays >50-fold selectivity versus the P2Y4 (EC50 of 9.3 μM) and P2Y6 (EC50 of 7.0 μM) receptors .
|
-
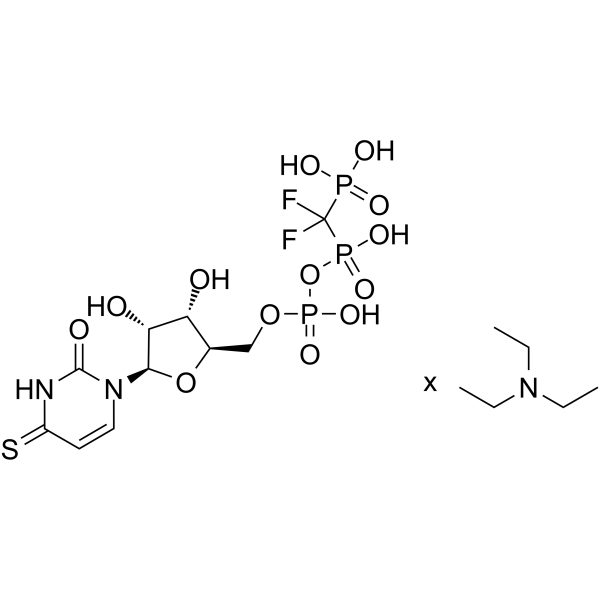
- HY-P1221A
-
|
|
Sodium Channel
|
Neurological Disease
|
|
ProTx II TFA is a selective blocker of Nav1.7 sodium channels with an IC50 of 0.3 nM, and is at least 100-fold selective for Nav1.7 over other sodium channel subtypes. ProTx-II inhibits sodium channels by decreasing channel conductance and shifting activation to more positive potentials and blocks action potential propagation in nociceptors .
|
-

- HY-14342A
-
|
|
Others
|
Metabolic Disease
|
|
(R)-MK-5046 is the isomer of MK-5046 (HY-14342), and can be used as an experimental control. MK-5046 is a potent, selective and orally active Bombesin receptor subtype-3 (BRS-3) allosteric agonist with an IC50 and an EC50 value of 27 and 25 nM for hBRS-3, respectively. MK-5046 inhibits food intake and reduces body weight of diet-induced obese (DIO) mouse models. MK-5046 can be used for the research of obesity .
|
-
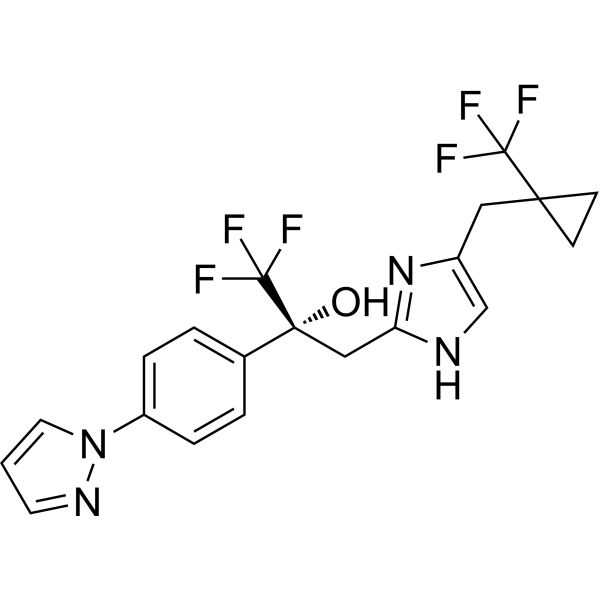
- HY-103187
-
|
|
Adenosine Receptor
|
Inflammation/Immunology
Cancer
|
|
HEMADO is a potent and selective adenosine A3 receptor agonist with a Ki of 1.1 nM at the human A3 subtype . HEMADO is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
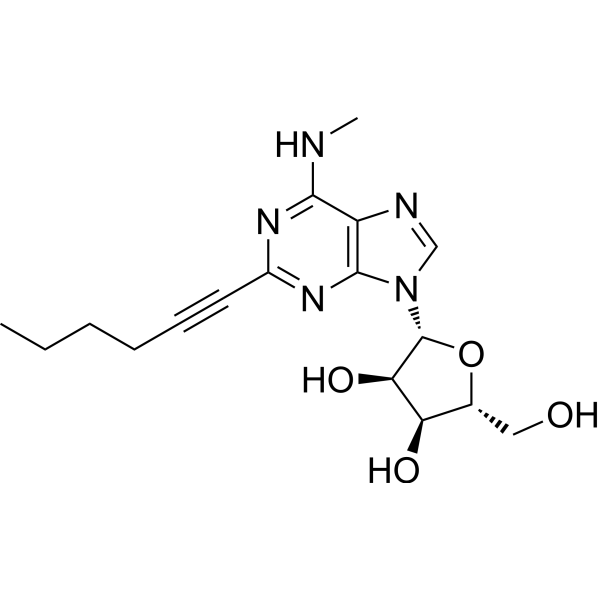
- HY-101079
-
|
|
|
|
|
BRL 52537 hydrochloride is a highly selective κ-Opioid receptor (KOR) agonist with Kis of 0.24 nM and 1560 nM for κ and μ subtypes, respectively. BRL 52537 hydrochloride decreases ischemia-evoked NO production as a potential mechanism of neuroprotection. BRL 52537 hydrochloride attenuates early stroke damage .
|
-
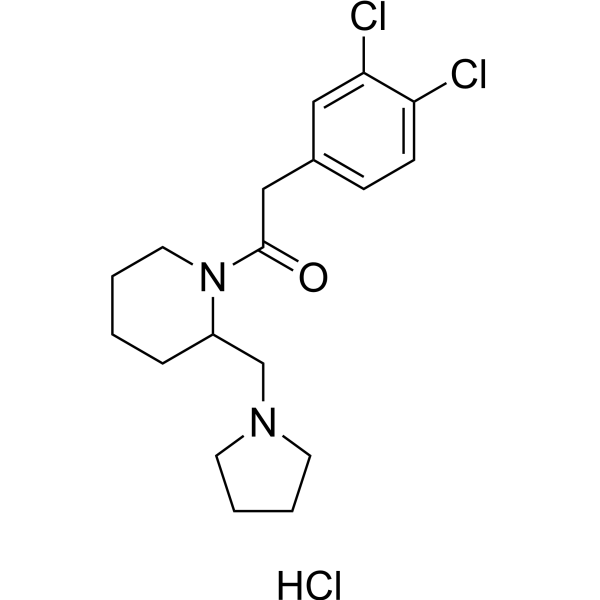
- HY-13225A
-
|
RJR-2403; (E)-Metanicotine
|
nAChR
|
Neurological Disease
|
|
Rivanicline (RJR-2403; (E)-Metanicotine) is a neuronal nicotinic receptor (neuronal nicotinic receptor) agonist that is highly selective for the α4β2 subtype,Ki is 26 nM, which is more than 1000 times more inhibitory than α7 receptor .
|
-
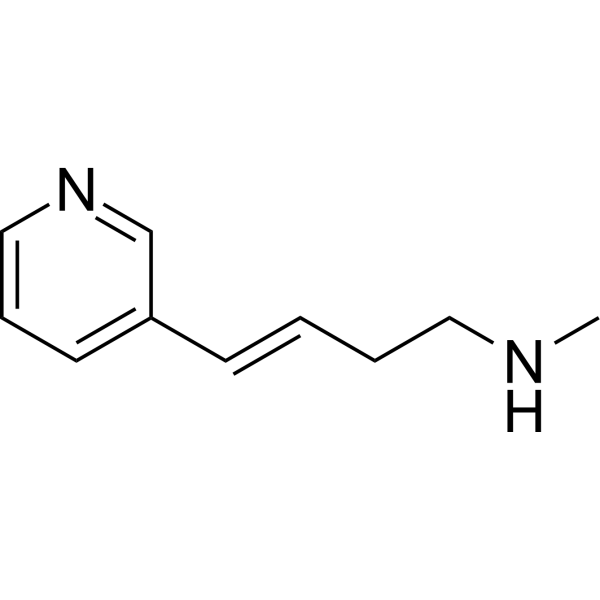
- HY-109085
-
|
OPA-15406
|
Phosphodiesterase (PDE)
|
Inflammation/Immunology
|
|
Difamilast (OPA-15406) is a topical, selective and nonsteroidal phosphodiesterase-4 (PDE4) inhibitor with particularly efficient inhibition of subtype B (IC50=11.2 nM). Difamilast can be used for the research of mild to moderate atopic dermatitis (AD) .
|
-
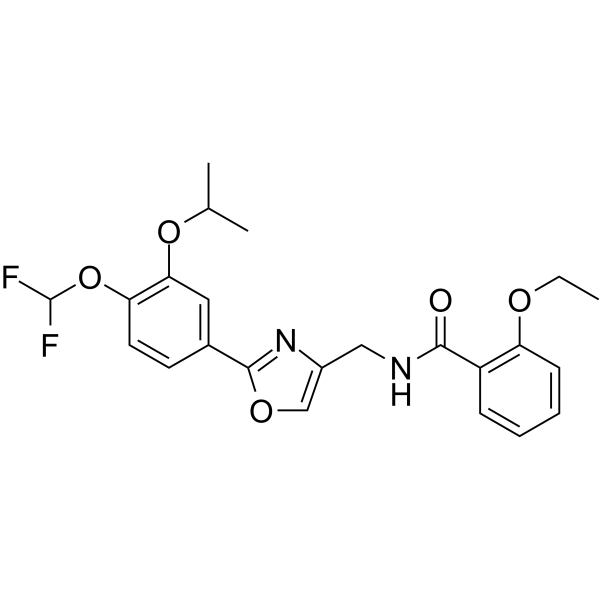
- HY-11048
-
NS11394
3 Publications Verification
|
GABA Receptor
|
Neurological Disease
|
|
NS11394 is an orally active and unique subtype-selective GABAA positive allosteric receptor (PAM), with a Ki of ~0.5 nM. NS11394 shows a selectivity profile in the order of GABAA-5 > α3 > α2 > α1-containing receptors. NS11394 has anxiolytic and anti-inflammatory properties .
|
-
- HY-122560A
-
|
|
Potassium Channel
|
Cardiovascular Disease
|
|
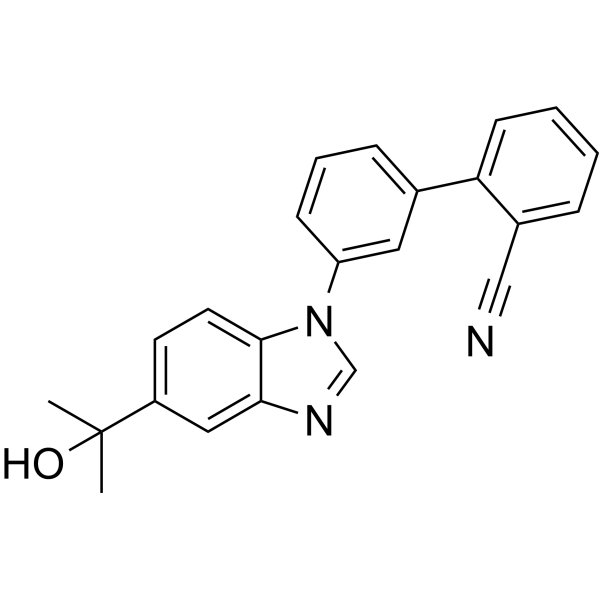
VU0134992 hydrochloride is the first subtype-preferring, orally active and selective Kir4.1 potassium channel pore blocker, with an IC50 of 0.97 µM. VU0134992 hydrochloride is 9-fold selective for homomeric Kir4.1 over Kir4.1/5.1 concatemeric channels (IC50=9 µM) at -120 mV .
|
-
- HY-122560
-
|
|
Potassium Channel
|
Cardiovascular Disease
|
|
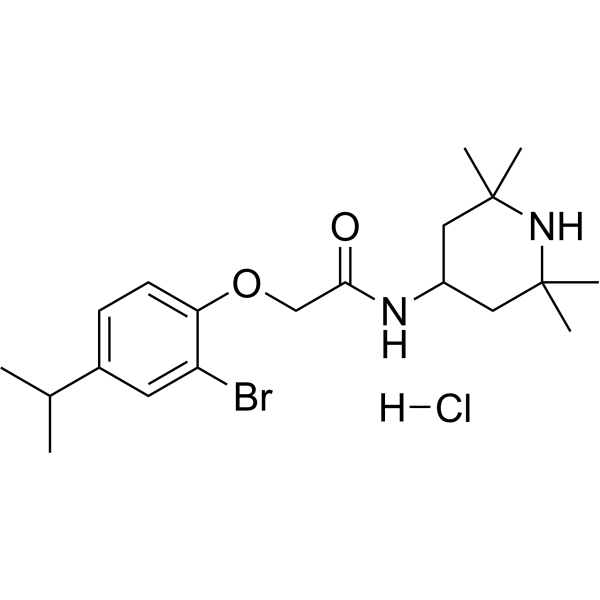
VU0134992 is the first subtype-preferring, orally active and selective Kir4.1 potassium channel pore blocker, with an IC50 of 0.97 µM. VU0134992 is 9-fold selective for homomeric Kir4.1 over Kir4.1/5.1 concatemeric channels (IC50=9 µM) at -120 mV .
|
-
- HY-107628
-
|
|
Melatonin Receptor
|
Neurological Disease
|
|
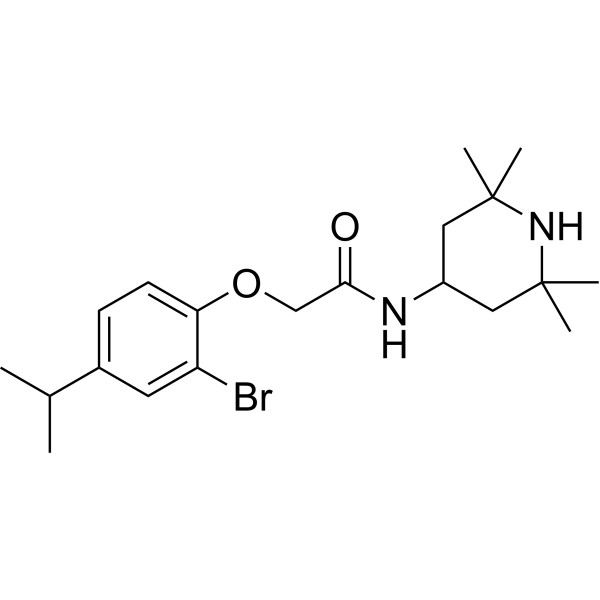
DH97 is a potent and selective antagonist of MT2 melatonin receptor, with a pKi of 8.03 for human MT2. DH97 shows 89- and 229-fold selectivity for human MT2 over human mt1 and Xenopus mel1c receptor subtypes. DH97 can inhibit melatonin-induced enhancement of electrically-evoked responses .
|
-
- HY-P5010
-
|
|
Vasopressin Receptor
|
Cardiovascular Disease
|
|
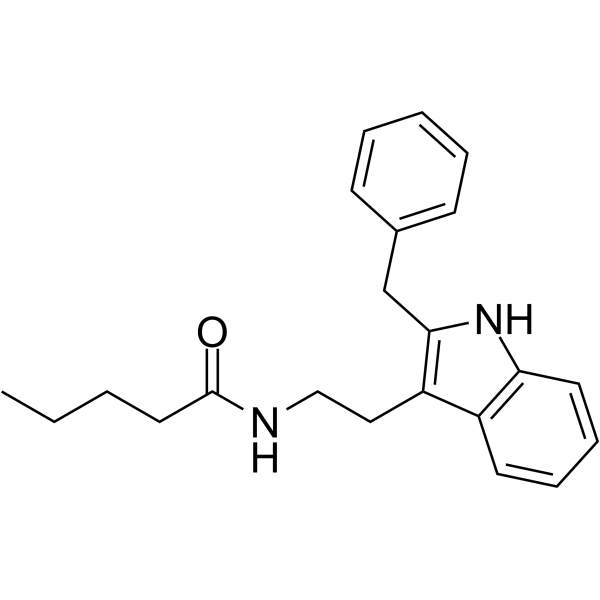
(D-Arg8)-Inotocin is a potent, selective and competitive antagonist of vasopressin receptor (V1aR), with a Ki of 1.3 nM. (D-Arg8)-Inotocin shows more than 3000-fold selectivity for the human V1aR over the other three subtypes, OTR, V1bR and V2R .
|
-
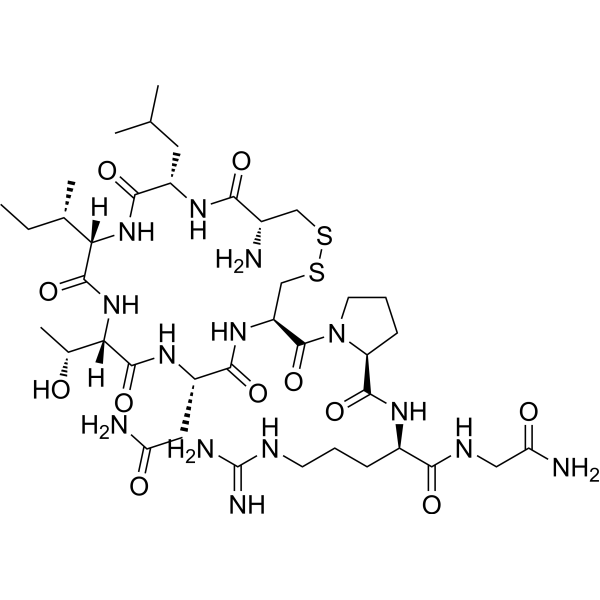
- HY-14567
-
|
FUB-359
|
Histamine Receptor
|
Neurological Disease
Endocrinology
|
|
Ciproxifan (FUB 359) is a potent, selective, orally bioavailable and competitive antagonist of histamine H3-receptor, with an IC50 of 9.2 nM. Ciproxifan displays low apparent affinity at other receptor subtypes. Ciproxifan can be used for the research of aging disorders and Alzheimer's disease .
|
-
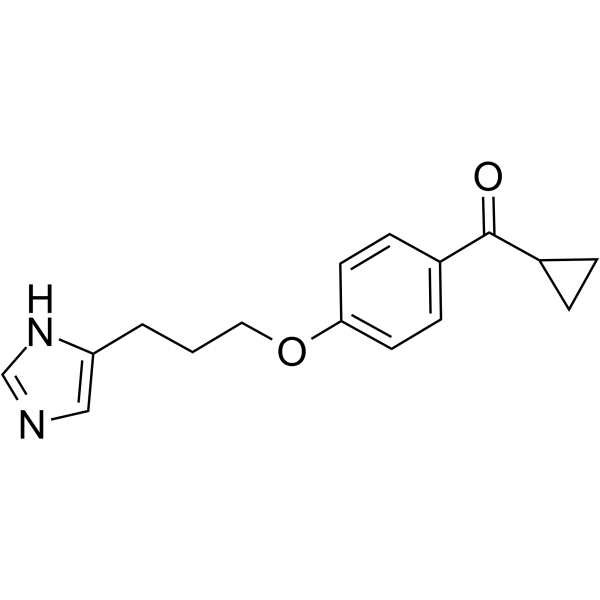
- HY-15289
-
|
FUB 359 maleate
|
Histamine Receptor
|
Neurological Disease
Endocrinology
|
|
Ciproxifan maleate (FUB 359 maleate) is a potent, selective, orally bioavailable and competitive antagonist of histamine H3-receptor, with an IC50 of 9.2 nM. Ciproxifan maleate displays low apparent affinity at other receptor subtypes. Ciproxifan maleate can be used for the research of aging disorders and Alzheimer's disease .
|
-
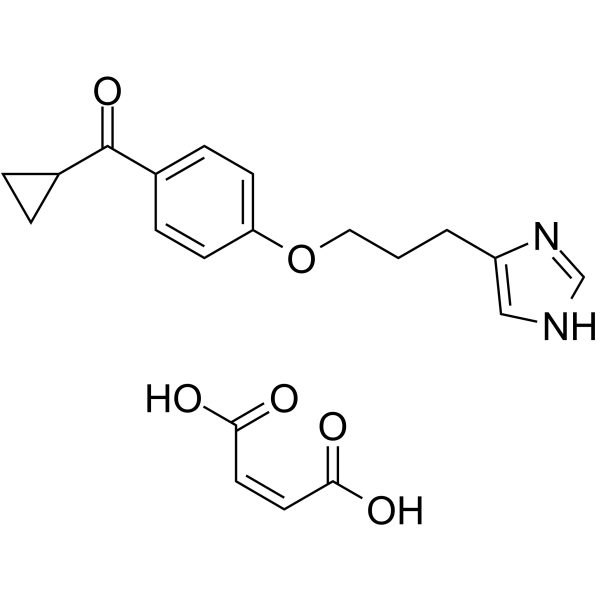
- HY-131973
-
|
|
Phosphodiesterase (PDE)
|
Cardiovascular Disease
Metabolic Disease
|
|
PDE10A-IN-2 hydrochloride is a potent, highly selective and orally active phosphodiesterase 10A (PDE10A) inhibitor with an IC50 of 2.8 nM. PDE10A-IN-2 hydrochloride shows selectivity of >3500-fold against other PDE subtypes. PDE10A-IN-2 hydrochloride can be used for pulmonary arterial hypertension (PAH) research .
|
-
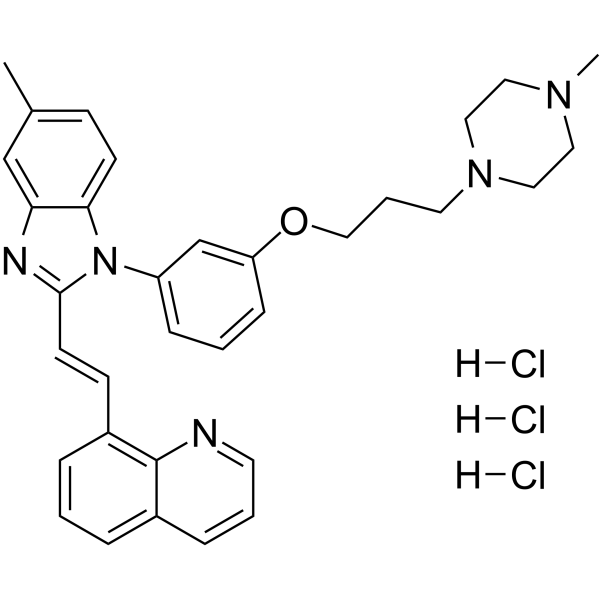
- HY-152947
-
|
|
Others
|
Metabolic Disease
|
|
ELOVL6-IN-4 is a potent, selective, and orally active long chain fatty acid elongase 6 (ELOVL6) inhibitor with IC50s of 79 nM and 94 nM for human and mouse ELOVL6, respectively. ELOVL6-IN-4 shows excellent selectivity over the other human ELOVL subtypes (ELOVL1, -2, -3, and -5) and mouse ELOVL3 .
|
-
- HY-111242
-
|
|
Dopamine Receptor
|
Neurological Disease
|
|
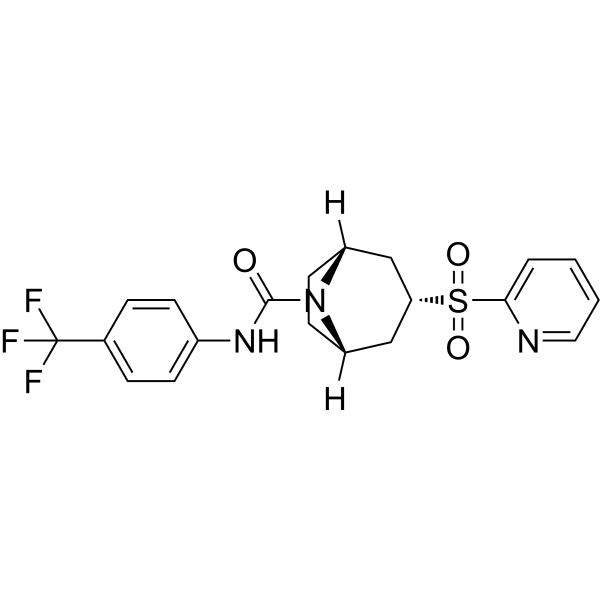
SV 156 is a potent and selective D2 dopamine receptor antagonist, with a Ki of 2.5 nM for hD2. SV 156 has approximately 40-fold binding selectivity for D2 dopamine receptors compared to the D3 receptor subtype. SV 156 can be used for L-DOPA (HY-N0304)-associated abnormal involuntary movements (AIMs) research .
|
-
- HY-110146
-
|
|
mGluR
|
Neurological Disease
|
|
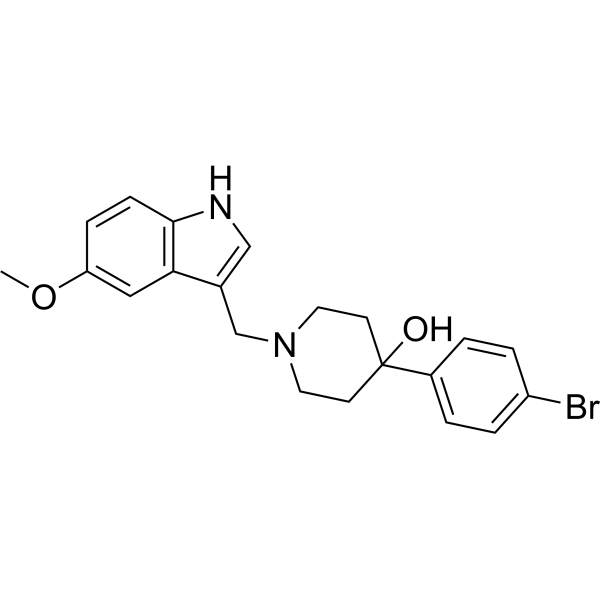
XAP044 is a potent and selective antagonist of mGlu7. The metabotropic glutamate receptor subtype 7 (mGlu7) is an important presynaptic regulator of neurotransmission in the mammalian CNS. XAP044 demonstrates good brain exposure and wide spectrum anti-stress and antidepressant- and anxiolytic-like efficacy in rodent behavioral paradigms .
|
-
- HY-119226
-
|
|
mAChR
|
Neurological Disease
|
|
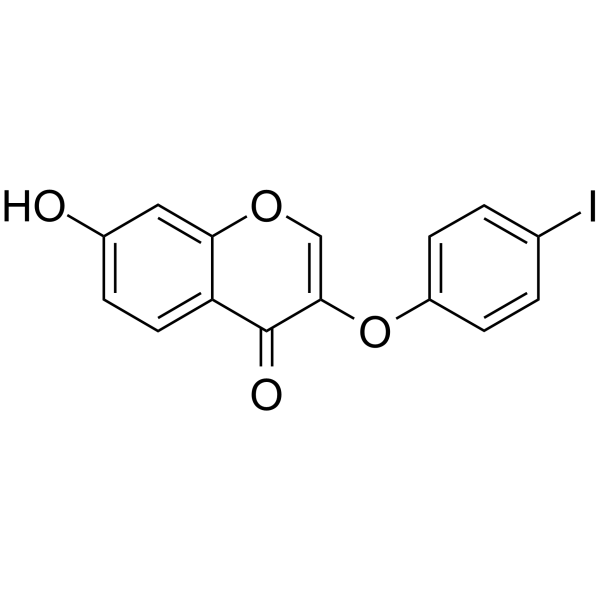
VU0152099 is a potent, selective and brain-penetrant mAChR M4 positive allosteric modulator with an EC50 of 0.4 µM for rat M4 receptor. VU0152099 is inactive for other mAChR subtypes or other GPCRs. VU0152099 has no agonist activity but potentiated responses of M4 to acetylcholine .
|
-
- HY-152227
-
|
|
PROTACs
JAK
|
Inflammation/Immunology
|
|
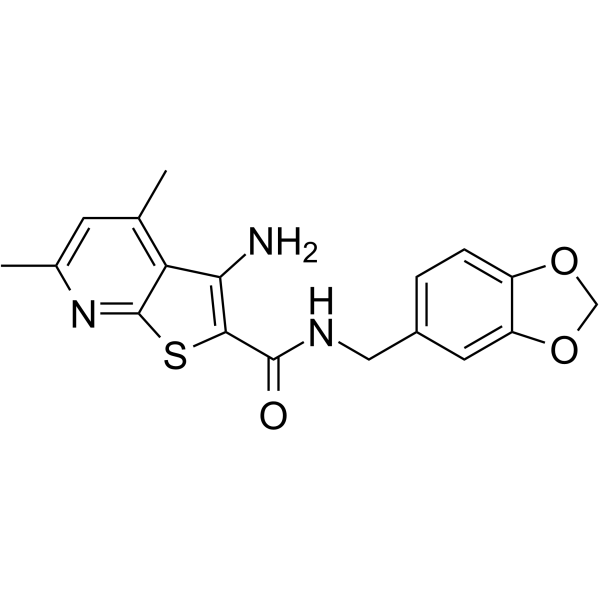
PROTAC TYK2 degradation agent1 is a potent and subtype-selective TYK2 degrader. PROTAC TYK2 degradation agent1 has TYK2 degradation activity with DC50 value of 14 nM. PROTAC TYK2 degradation agent1 can be used for the research of autoimmune disease .
|
-
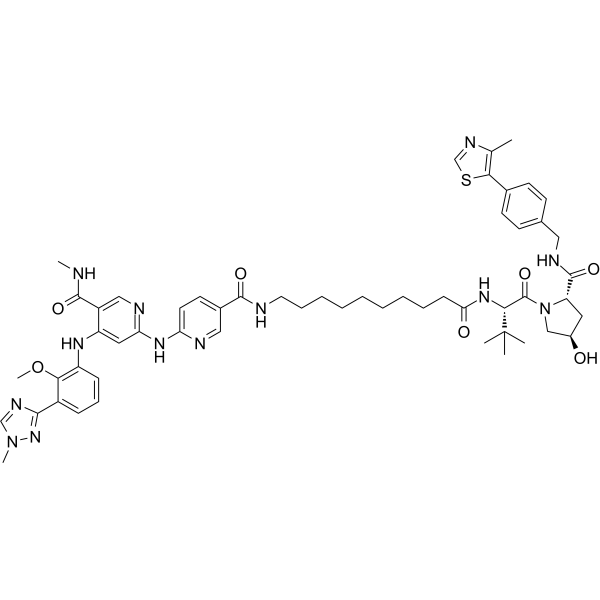
- HY-108669
-
|
|
P2X Receptor
|
Neurological Disease
Cancer
|
|
AZ10606120 dihydrochloride is a selective, high affinity antagonist for P2X7 receptor (P2X7R) at human and rat with an IC50 of about 10 nM. AZ10606120 dihydrochloride is little or no effect at other P2XR subtypes. AZ10606120 dihydrochloride has anti-depressant effects and reduces tumour growth .
|
-
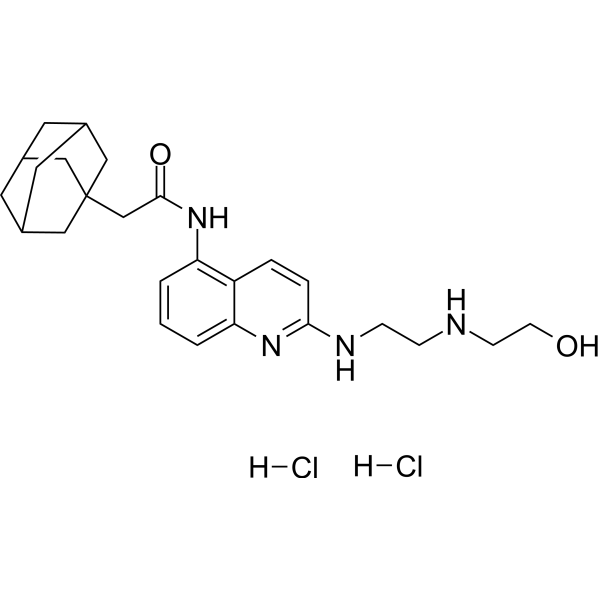
- HY-147730
-
|
|
HDAC
|
Cancer
|
|
A variety of compounds were designed and synthesized by modifying cap groups. The enzyme inhibition test showed that compound 12C had broad-spectrum enzyme inhibitory activity, and compounds 9m and 9q were more inclined to inhibit HDAC6, showing a certain selective inhibitory activity among the representative subtypes.
|
-
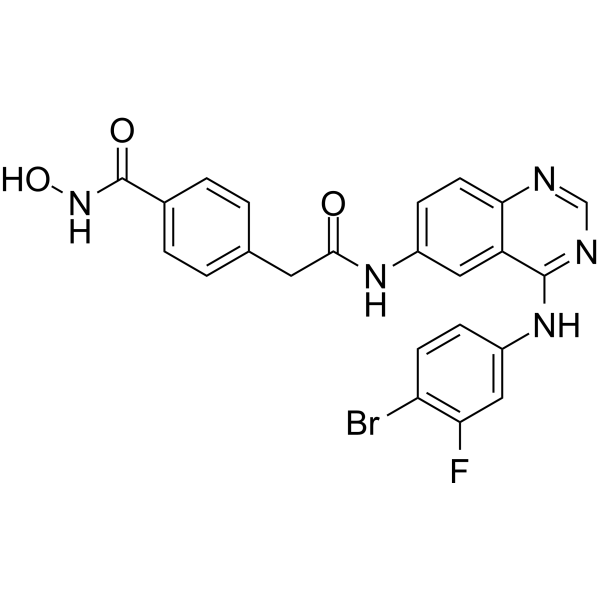
- HY-151487
-
|
|
Sirtuin
|
Neurological Disease
Metabolic Disease
Cancer
|
|
CypD-IN-3 is a potent and subtype-selective cyclophilin D (CypD) inhibitor. CypD-IN-3 has CypD affinity with an IC50 value of 0.01 μM. CypD-IN-3 can be used for the research of several diseases including oxidative stress, neurodegenerative disorders, liver diseases, aging, autophagy and diabetes .
|
-
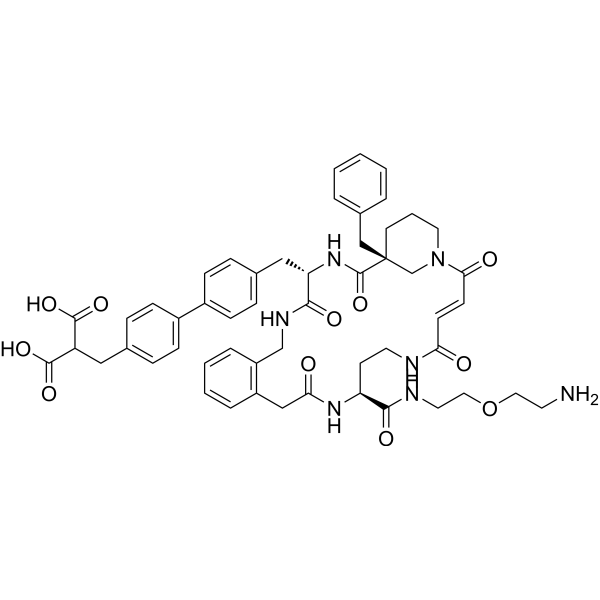
- HY-151488
-
|
|
Sirtuin
|
Neurological Disease
Metabolic Disease
Cancer
|
|
CypD-IN-4 is a potent and subtype-selective cyclophilin D (CypD) inhibitor. CypD-IN-4 has CypD affinity with an IC50 value of 0.057 μM. CypD-IN-4 can be used for the research of several diseases including oxidative stress, neurodegenerative disorders, liver diseases, aging, autophagy and diabetes .
|
-
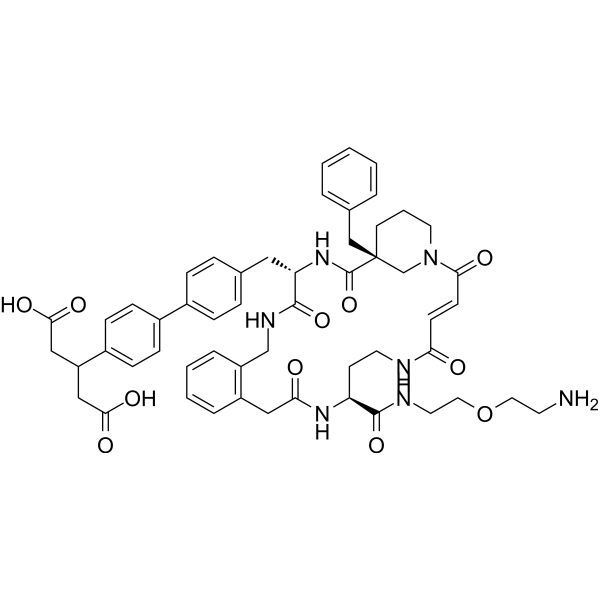
- HY-128686
-
KAG-308
1 Publications Verification
|
Prostaglandin Receptor
|
Inflammation/Immunology
Endocrinology
|
|
KAG-308 is a potent selective and orally active agonist of EP4 receptor (a prostaglandin E2 receptor subtype), suppresses colitis and promotes histological mucosal healing, potently inhibits TNF-α production. KAG-308 shows a Ki and an EC50 of 2.57 nM and 17 nM for human EP4 receptor, respectively, more selective over EP1, EP2, EP3 and IP receptor .
|
-
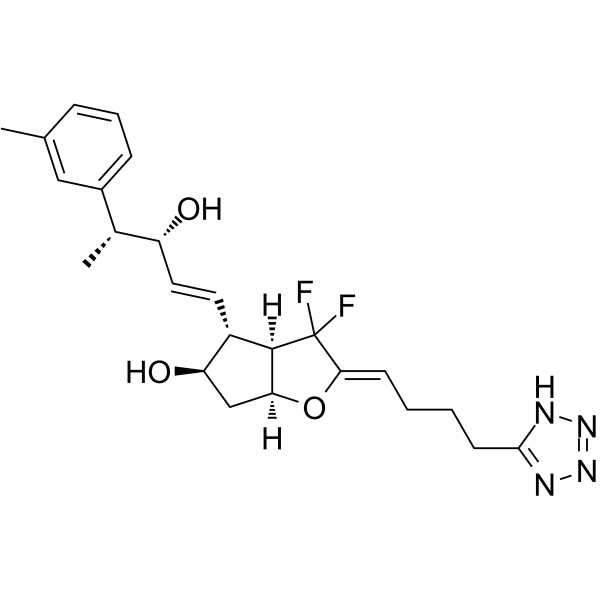
- HY-103565
-
|
|
mGluR
|
Neurological Disease
|
|
AMN082, a selective, orally active, and brain penetrant mGluR7 agonist, directly activates receptor signaling via an allosteric site in the transmembrane domain. AMN082 potently inhibits cAMP accumulation and stimulates GTPγS binding (EC50 values, 64-290 nM) at transfected mammalian cells expressing mGluR7. AMN082 shows selectivity over other mGluR subtypes and selected ionotropic glutamate receptors. Antidepressant effects .
|
-
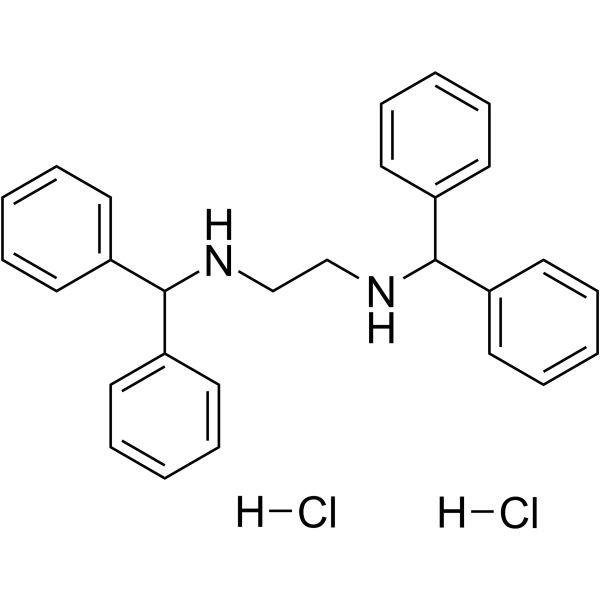
- HY-152576
-
|
|
Cannabinoid Receptor
|
Inflammation/Immunology
|
|
CB2R agonist 1 is a selective ligand of cannabinoid receptor subtype 2 (CB2R) with an EC50 value of 0.56 µM. CB2R agonist 1 has high affinity and excellent selectivity for human CB2R and CB1R respectively. CB2R agonist 1 regulates the production of pro-inflammatory cytokines and anti-inflammatory cytokines and play an immunomodulatory role .
|
-
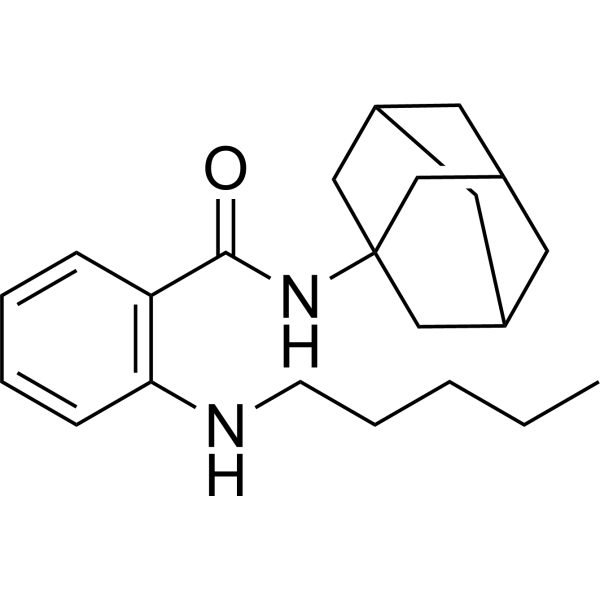
- HY-111501
-
|
|
Histamine Receptor
|
Inflammation/Immunology
Endocrinology
|
|
H4R antagonist 1 is a potent and highly selective histamine H4 receptor (H4R) antagonist with an IC50 of 27 nM. H4R antagonist 1 does not show any noticeable binding affinity to other subtypes of histamine receptors, H1R, H2R, and H3R .
|
-
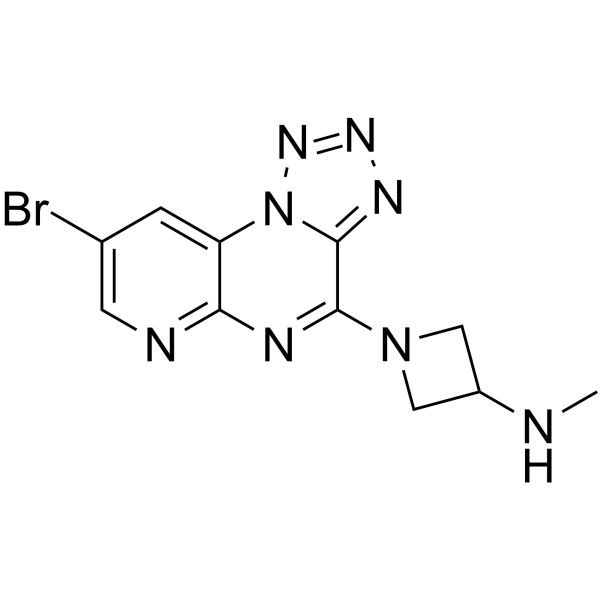
- HY-111539
-
|
|
Prostaglandin Receptor
|
Inflammation/Immunology
Endocrinology
|
|
BAY-1316957 is a potent, selective and orally active prostaglandin E2 receptor subtype 4 (EP4-R) antagonist with an IC50 of 15.3 nM for human EP4-R. BAY-1316957 has excellent agent metabolism and pharmacokinetics properties, and can be used for endometriosis research .
|
-
- HY-17416A
-
|
|
Adrenergic Receptor
|
Neurological Disease
Endocrinology
|
|
Guanfacine is an orally active noradrenergic α2A agonist and has high selective for the α2A receptor subtype. Guanfacine has effects in producing hypotension and sedation. Guanfacine can be used for the research of a variety of prefrontal cortex (PFC) cognitive disorders, including tourette's syndrome and attention deficit hyperactivity disorder (ADHD) .
|
-
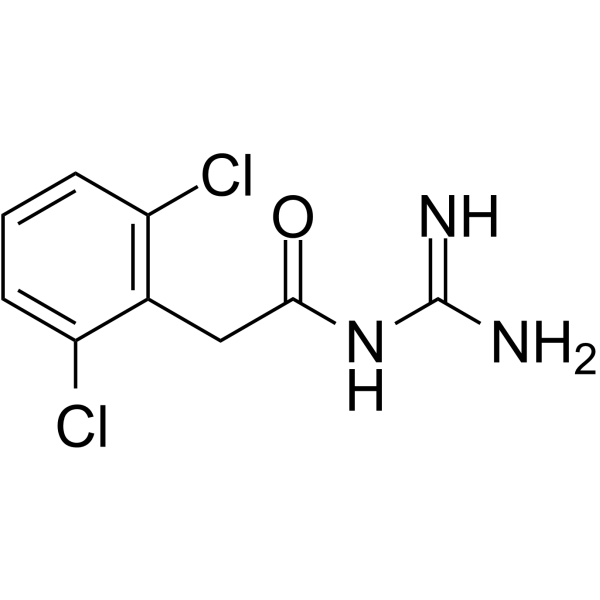
- HY-17416
-
|
|
Adrenergic Receptor
|
Cardiovascular Disease
Endocrinology
|
|
Guanfacine hydrochloride is an orally active noradrenergic α2A agonist and has high selective for the α2A receptor subtype. Guanfacine has effects in producing hypotension and sedation. Guanfacine can be used for the research of a variety of prefrontal cortex (PFC) cognitive disorders, including tourette's syndrome and attention deficit hyperactivity disorder (ADHD) .
|
-
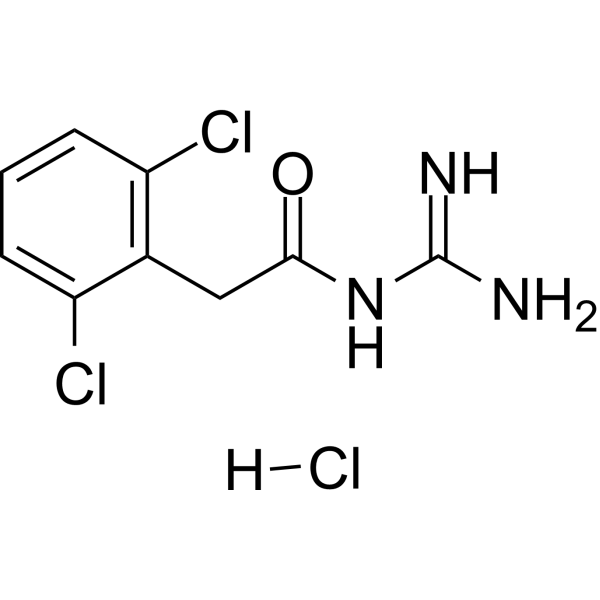
- HY-15023
-
|
PF-3274167
|
Oxytocin Receptor
|
Endocrinology
|
|
Cligosiban (PF-3274167), a high oral bioavailability and good brain-penetrant non-peptide oxytocin receptor antagonist, shows a high-affinity (Ki=9.5 nM) and an excellent selectivity versus the vasopressin receptors with almost no affinity for the V1b and V1a subtypes. Cligosiban inhibits ejaculatory physiology in rodents .
|
-
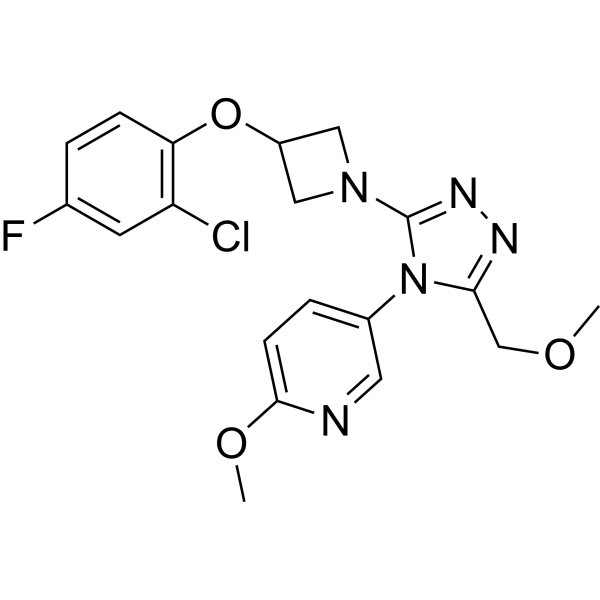
- HY-151489
-
|
|
Sirtuin
|
Neurological Disease
Metabolic Disease
Cancer
|
|
CypD-IN-1 is a potent and subtype-selective cyclophilin E (CypE) inhibitor. CypD-IN-1 has CypE affinity with IC50 and Ki values of 0.013 μM and 0.072 μM, respectively. CypD-IN-1 can be used for the research of several diseases including oxidative stress, neurodegenerative disorders, liver diseases, aging, autophagy and diabetes .
|
-
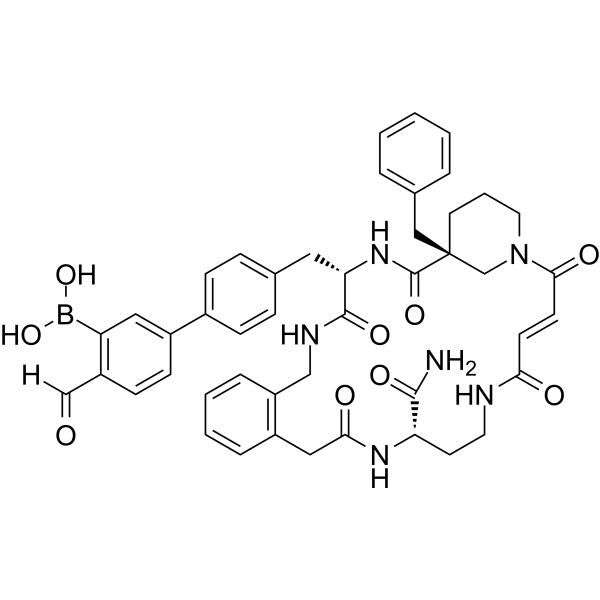
- HY-P3676
-
|
|
Neuropeptide Y Receptor
|
Neurological Disease
|
|
Neuropeptide Y (3-36) (porcine) is an agonist of neuropeptide Y (NPY) receptor subtype Y2, and stimulates feeding in rats. Neuropeptide Y (3-36) (porcine) is a highly Y2 selective ligand compared with nselective Y1/Y2 receptor ligand, Neuropeptide Y 1-36 .
|
-

- HY-126291
-
|
|
Sodium Channel
|
Neurological Disease
|
|
GNE-616 is a highly potent, metabolically stable, orally bioavailable, and subtype selective Nav1.7 inhibitor (Ki of 0.79 nM and Kd of 0.38 nM for hNav1.7) for the treatment of chronic pain. GNE-616 shows >1000 nM Kd and >2500-fold selectivity over hNav1.1, hNav1.3, hNav1.4, and hNav1.5. Selectivity over hNav1.2 and hNav1.6 is more modest at 31- and 73-fold, respectively .
|
-
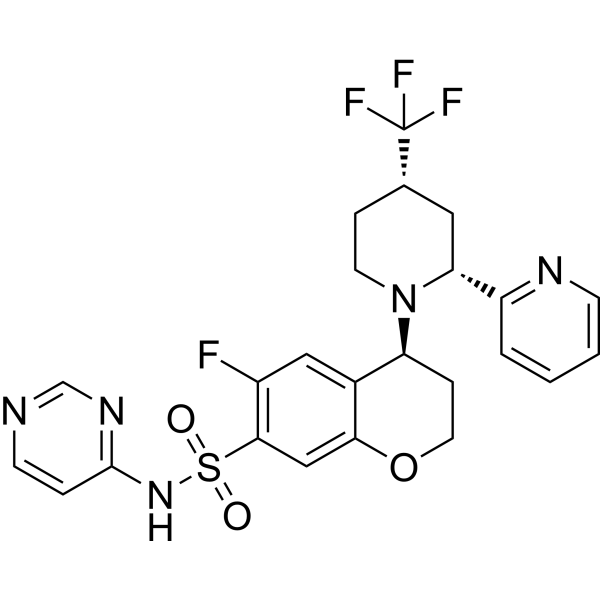
- HY-111557
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
YM-254890 is a selective Gαq/11 protein inhibitor isolated from Chromobacterium sp. YM-254890 shows no inhibition of other G protein subtypes. YM-254890 inhibits platelet aggregation induced by ADP by blocking the P2Y1 signal transduction pathway, with an IC50 value below 0.6 μM .
|
-
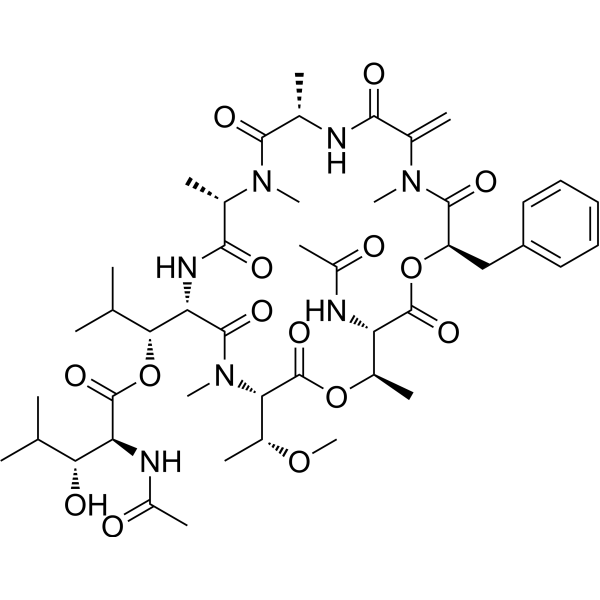
- HY-103212
-
|
B-HT 933 dihydrochloride; Oxazoloazepin dihydrochloride
|
Adrenergic Receptor
|
Cardiovascular Disease
Endocrinology
|
|
Azepexole (B-HT 933) dihydrochloride is a potent and selective alpha 2-adrenoceptor agonist with pKis of 8.3, 7.6, and 7.5 for α2A-, α2B- and α2C-adrenoceptor subtypes, resepctively . Azepexole dihydrochloride causes concentration-dependent inhibition of peristaltic contractions (IC50= 78.72 nM) .
|
-
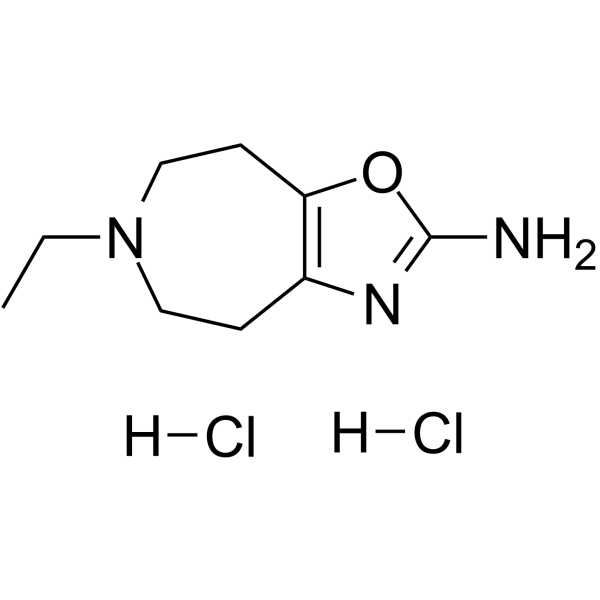
- HY-103565A
-
|
|
mGluR
|
Neurological Disease
|
|
AMN082 free base, a selective, orally active, and brain penetrant mGluR7 agonist, directly activates receptor signaling via an allosteric site in the transmembrane domain. AMN082 free base potently inhibits cAMP accumulation and stimulates GTPγS binding (EC50 values, 64-290 nM) at transfected mammalian cells expressing mGluR7. AMN082 free base shows selectivity over other mGluR subtypes and selected ionotropic glutamate receptors. Antidepressant effects .
|
-
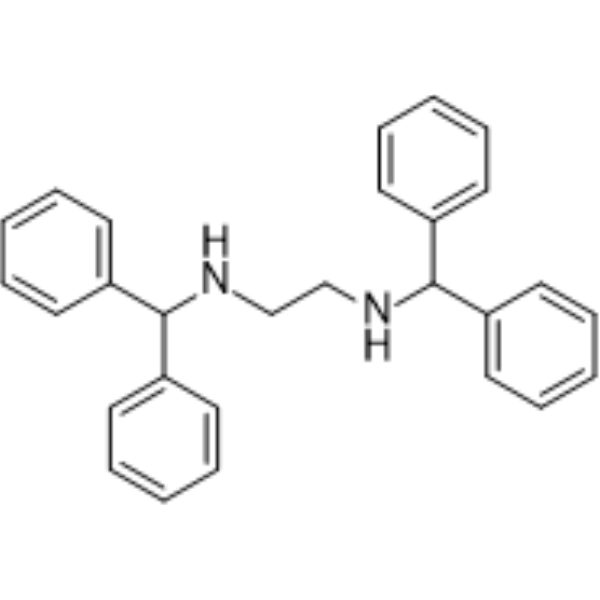
- HY-161306
-
|
|
HDAC
|
Cancer
|
|
ITF5924 (compound 1) is a potent and highly selective HDAC6 inhibitor with an IC50 of 7.7 nM. ITF5924 shows greater than 104-fold selectivity for HDAC6 over all other HDAC subtypes. ITF5924 containing a difluoromethyl-1,3,4-oxadiazole (DFMO) moiety is slow-binding substrate analog of HDAC6 that undergo an enzyme-catalyzed ring opening reaction, forming a tight and long-lived enzyme-inhibitor complex .
|
-
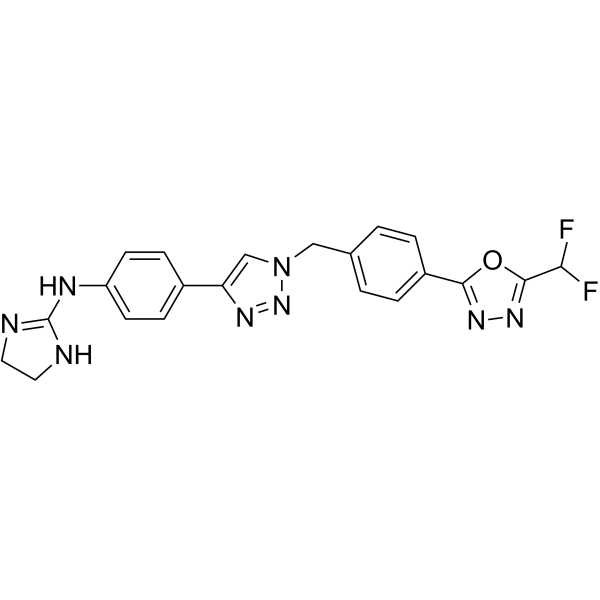
- HY-B0391B
-
|
KT-611 hydrochloride; BM-15275 hydrochloride
|
Adrenergic Receptor
|
Cancer
|
|
Naftopidil hydrochloride (KT-611 hydrochloride) is a selective alpha1-adrenoceptor antagonist, with Kis of 3.7 nM, 20 nM and 1.2 nM for the cloned human α1a-, α1b- and α1d-adrenoceptor subtypes, respectively. Naftopidil hydrochloride has antiproliferative effects. Naftopidil hydrochloride can be used for the research of prostate hyperplasia .
|
-
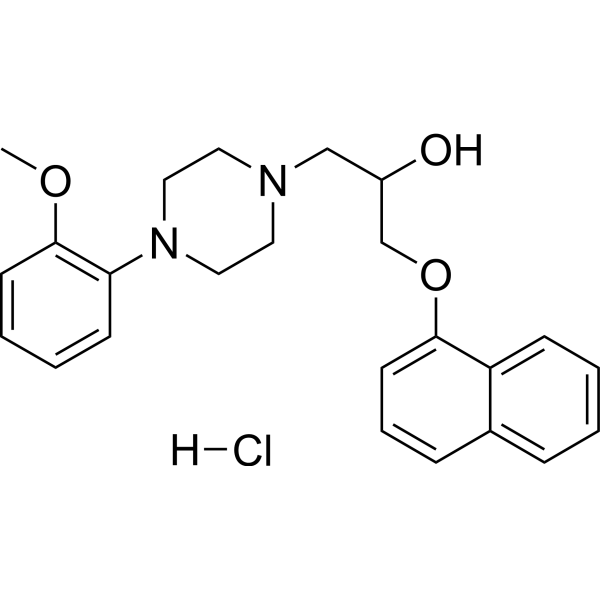
- HY-123249
-
|
|
GABA Receptor
|
Neurological Disease
|
|
HZ166 is a GABAA receptor subtype-selective benzodiazepine site agonist with preferential activity at α2- and α3-GABAA receptors. HZ166 shows anti-hyperalgesic effects . HZ166 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
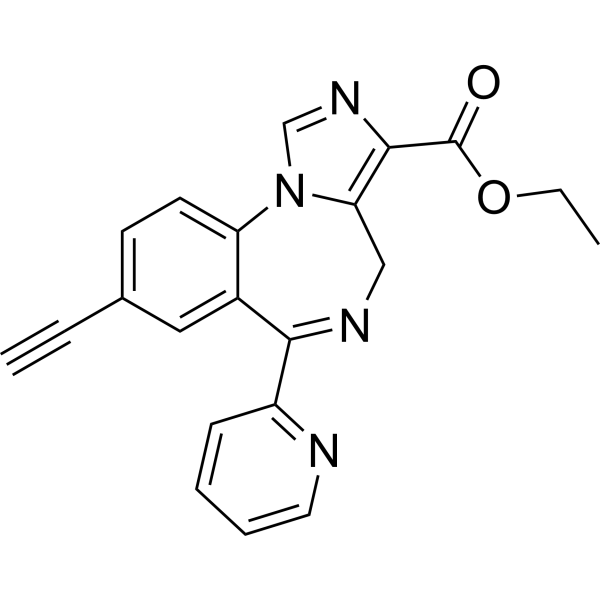
- HY-136207
-
|
|
nAChR
|
Neurological Disease
|
|
TC-2559 idifumarate is a CNS-selective, orally active α4β2 subtype of nicotinic acetylcholine receptor (nAChR) partial agonist (EC50=0.18 μM). TC-2559 difumarate shows selectivity for α4β2 over α2β4, α4β4 and α3β4 receptors, with EC50s in the range of 10-30 µM. Antinociceptive effect .
|
-
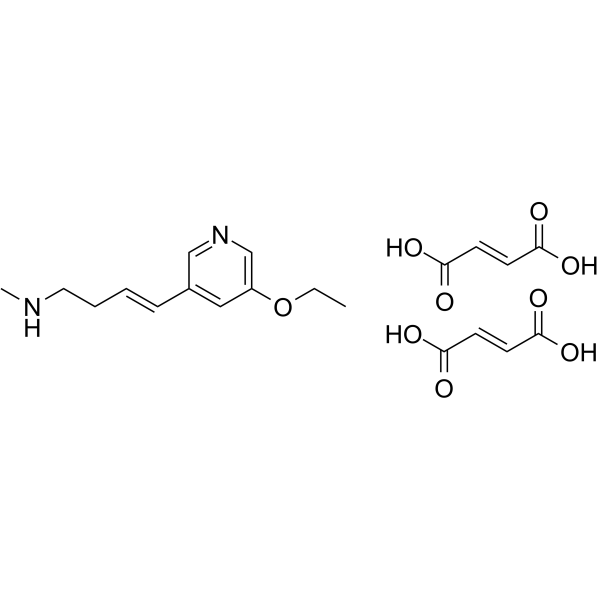
- HY-B0391
-
|
KT-611; BM-15275
|
Adrenergic Receptor
|
Cardiovascular Disease
Endocrinology
|
|
Naftopidil (KT-611) is is a selective alpha1-adrenoceptor antagonist, with Kis of 3.7 nM, 20 nM and 1.2 nM for the cloned human α1a-, α1b- and α1d-adrenoceptor subtypes, respectively. Naftopidil has antiproliferative effects. Naftopidil can be used for the research of prostate hyperplasia .
|
-
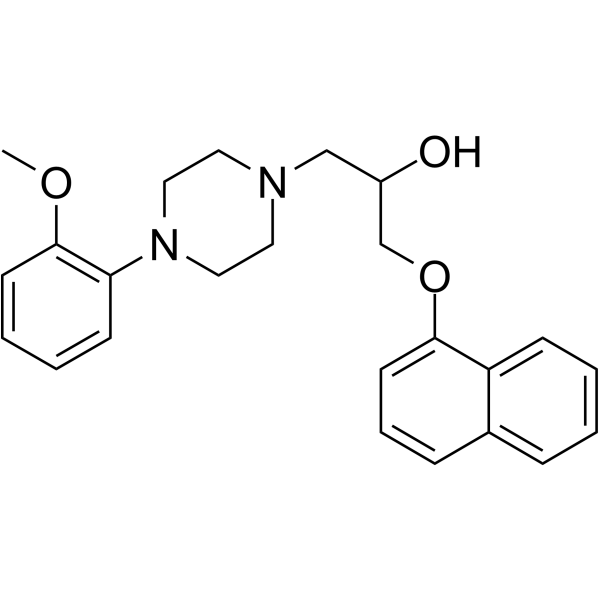
- HY-B0391A
-
|
KT-611 dihydrochloride; BM-15275 dihydrochloride
|
Adrenergic Receptor
|
Cancer
|
|
Naftopidil dihydrochloride (KT-611 dihydrochloride) is a selective alpha1-adrenoceptor antagonist, with Kis of 3.7 nM, 20 nM and 1.2 nM for the cloned human α1a-, α1b- and α1d-adrenoceptor subtypes, respectively. Naftopidil dihydrochloride has antiproliferative effects. Naftopidil dihydrochloride can be used for the research of prostate hyperplasia .
|
-
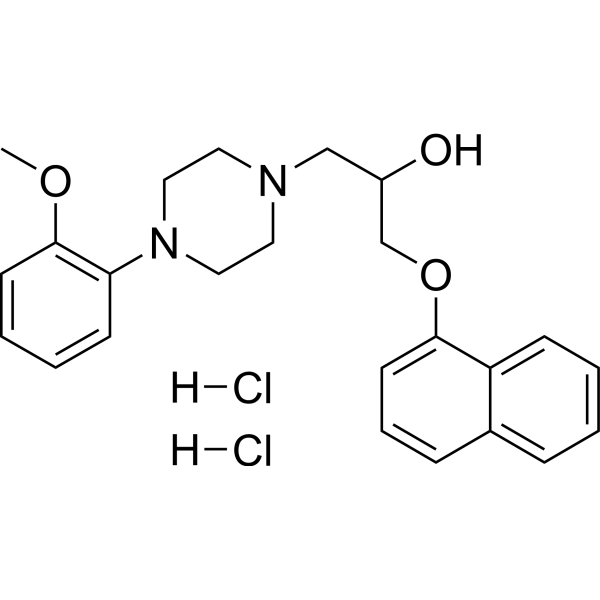
- HY-107508
-
|
|
mGluR
|
Neurological Disease
|
|
VU-29 is a positive allosteric modulator of metabotropic glutamate 5 (mGlu5) receptor (EC50=9 nM and Ki=244 nM for rmGluR5). VU-29 is selective for mGluR5 relative to other mGluR subtypes (EC50: rmGluR1/rmGluR2=557 nM/1.5 μM; hmGluR4=154 nM) .
|
-
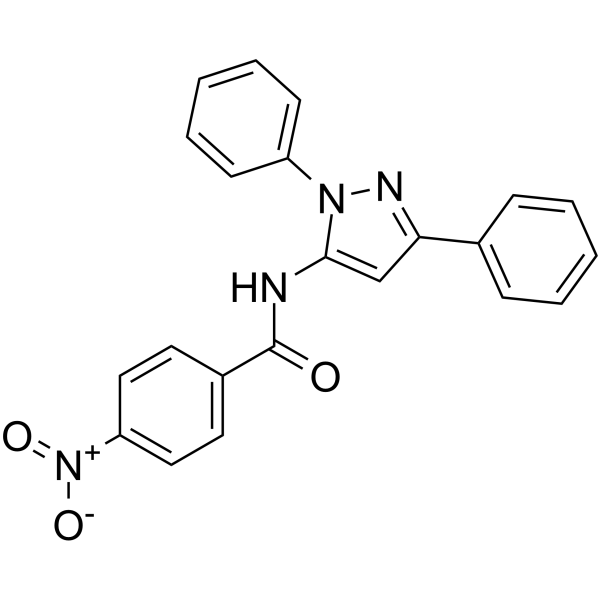
- HY-161405
-
|
|
Neuropeptide FF Receptor
Opioid Receptor
|
Neurological Disease
|
|
NPFF1-R antagonist 1 (compound 8b), a piperidine analogue, is a potent neuropeptide FF (NPFF) receptor antagonist. NPFF1-R antagonist 1 is 15-fold selective for the NPFF1-R subtype, with Ki values of 211 nM and 3270 nM for NPFF1-R and NPFF2-R, respectively .
|
-
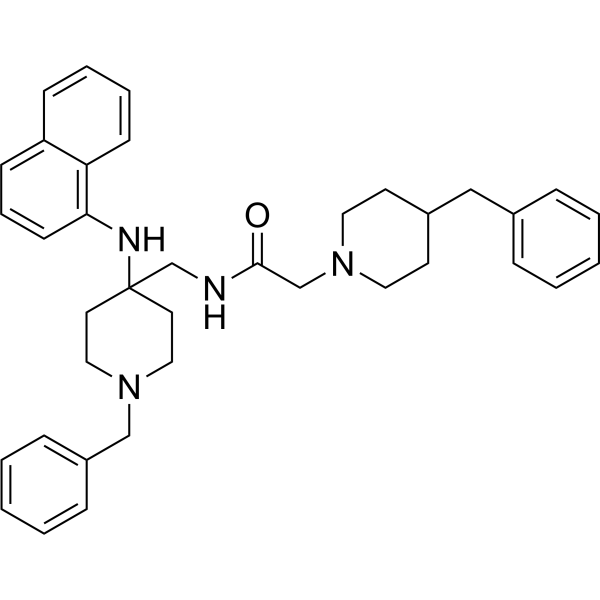
- HY-14350
-
|
|
Protease Activated Receptor (PAR)
|
Others
|
|
AC-55541 is a highly selective protease-activated receptor 2 (PAR2) agonist (pEC50=6.7), displays no activity at other PAR subtypes or at over 30 other receptors involved in nociception and inflammation. AC-55541 has pEC50 values of 5.9 and 6.6 in PI hydrolysis assays and Ca 2+ mobilization assays and exhibits pronociceptive activity in vivo .
|
-
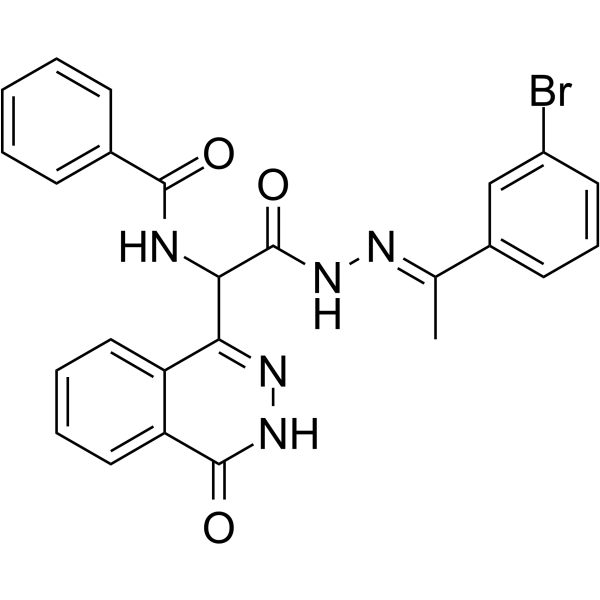
- HY-18596
-
|
|
5-HT Receptor
|
Neurological Disease
|
|
SB-215505 is a potent and subtype-selective 5-HT2B receptor antagonist with pKi values of 8.3, 6.77, 7.66 for 5-HT2B, 5-HT2A, 5-HT2C, respectively . SB-215505 increases wakefulness and motor activity in rats .
|
-
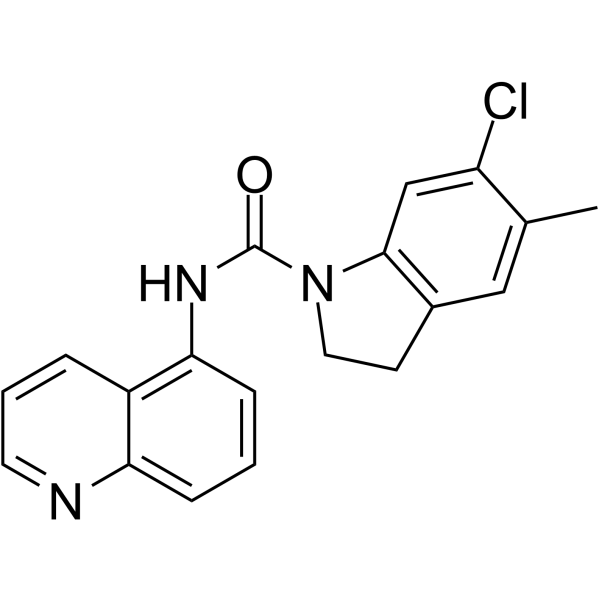
- HY-135483A
-
|
|
nAChR
|
Neurological Disease
|
|
AR-R17779 hydrochloride is a potent and selective full agonist of nAChR, with Kis of 92 and 16000 nM for α7 and α4β2 subtype, respectively. AR-R17779 hydrochloride can improve learning and memory in rats. AR-R17779 hydrochloride also has anxiolytic activity. AR-R17779 hydrochloride can reduce inflammation by activating antiinflammatory cholinergic (vagal) pathways .
|
-
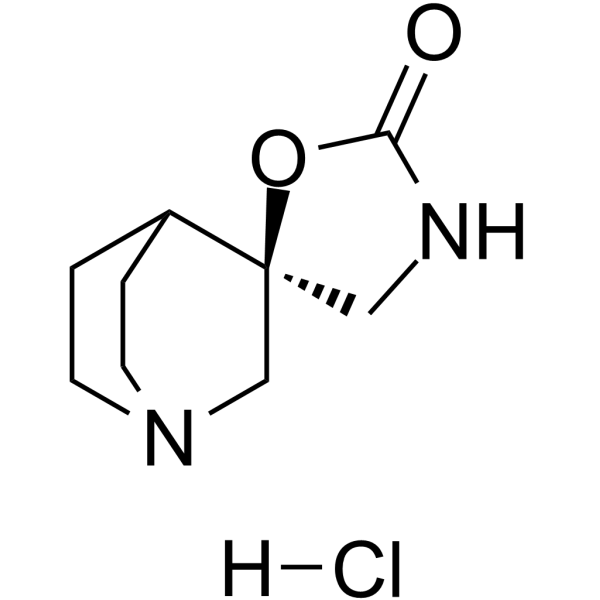
- HY-16787
-
|
|
Sodium Channel
|
Cardiovascular Disease
|
|
ICA-121431 is a nanomolar potent and broad-spectrum voltage-gated sodium channel (Nav) blocker, shows equipotent selectivity for human Nav1.1 and Nav1.3 subtypes with IC50 values of 13 nM and 23 nM, respectively. ICA-121431 shows less potent inhibition of Nav1.2 (IC50=240 nM) and 1,000 fold selectivity against Nav1.4, Nav1.6, and the TTX-resistant human Nav1.5 and Nav1.8 channels (IC50s >10 µM).
|
-
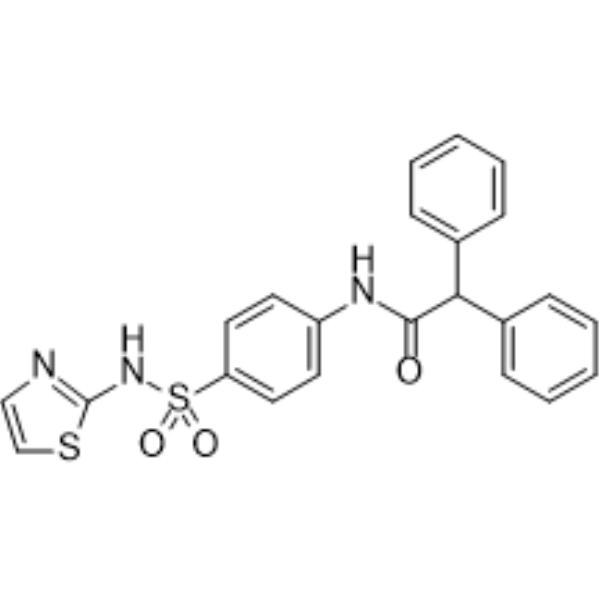
- HY-13285
-
|
Debio 0719
|
LPL Receptor
|
Neurological Disease
Cancer
|
|
Ki16425 (Debio 0719) is a subtype-selective, competitive antagonist of the EDG-family receptors, LPA1 and LPA3 with Kis of 0.34 μM and 0.93 μM, respectively. Ki16425 (Debio 0719) reduces the LPA-induced activation of p42/p44 MAPK . Ki16425 can also inhibit LPA-induced dephosphorylation of Yes-associated protein (YAP)/TAZ in HEK293A cells .
|
-
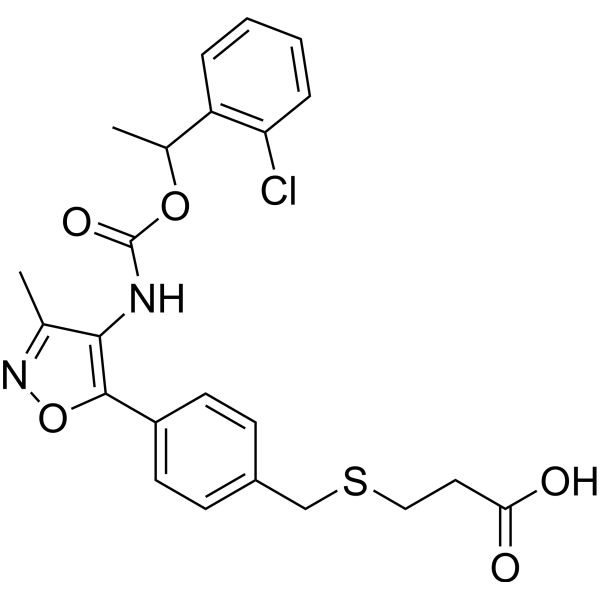
- HY-B0391S1
-
|
KT-611-d5; BM-15275-d5
|
Isotope-Labeled Compounds
Adrenergic Receptor
|
Cardiovascular Disease
Endocrinology
|
|
Naftopidil-d5 is deuterium labeled Naftopidil. Naftopidil (KT-611) is is a selective alpha1-adrenoceptor antagonist, with Kis of 3.7 nM, 20 nM and 1.2 nM for the cloned human α1a-, α1b- and α1d-adrenoceptor subtypes, respectively. Naftopidil has antiproliferative effects. Naftopidil can be used for the research of prostate hyperplasia[1][2].
|
-
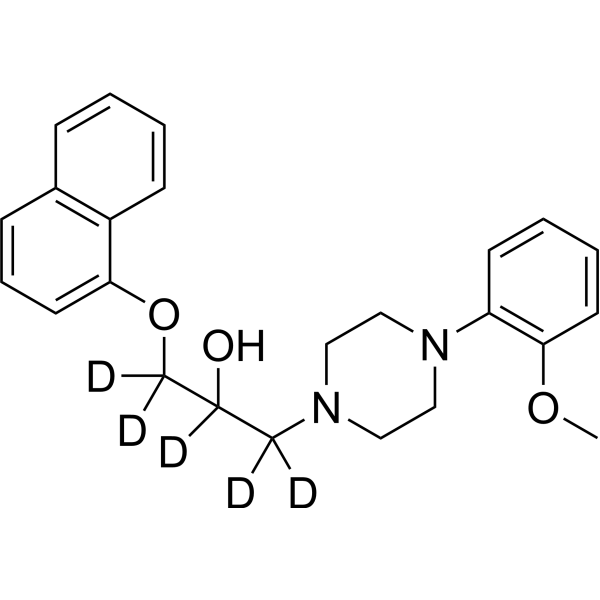
- HY-17416AS
-
|
|
Adrenergic Receptor
|
|
|
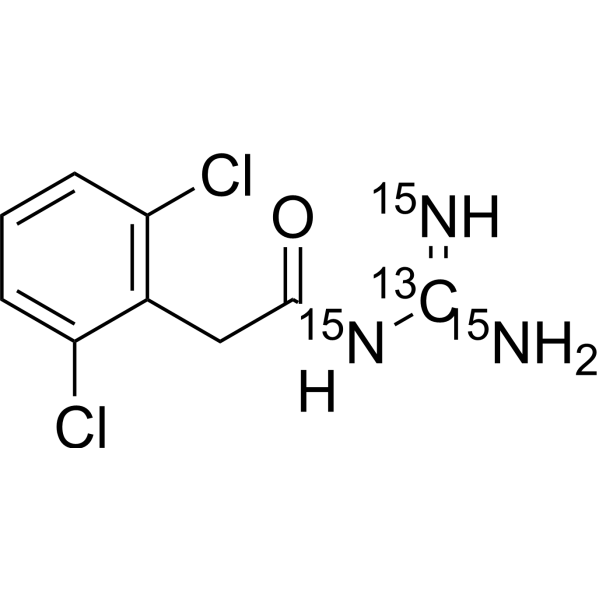
Guanfacine- 13C, 15N3 is the 13C and 15N labeled Guanfacine[1]. Guanfacine is an orally active noradrenergic α2A agonist and has high selective for the α2A receptor subtype. Guanfacine has effects in producing hypotension and sedation. Guanfacine can be used for the research of a variety of prefrontal cortex (PFC) cognitive disorders, including tourette's syndrome and attention deficit hyperactivity disorder (ADHD)[2][3][4].
|
-
- HY-17416S2
-
|
|
Adrenergic Receptor
Isotope-Labeled Compounds
|
Cardiovascular Disease
Endocrinology
|
|
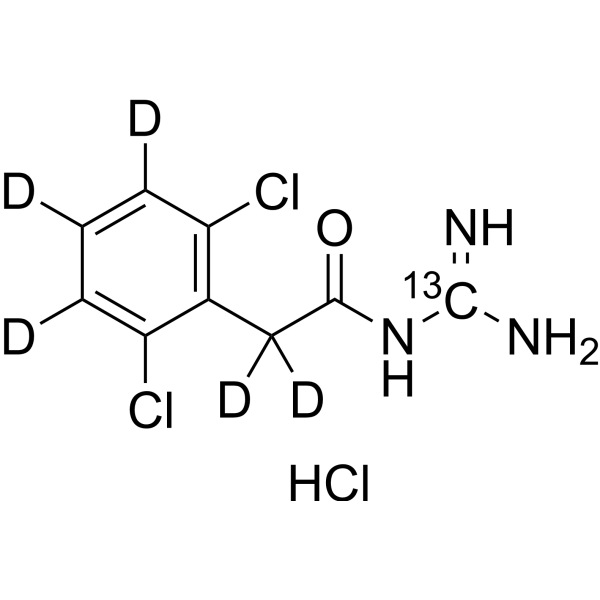
Guanfacine- 13C,d5 hydrochloride is the deuterium and 13C labeled Guanfacine hydrochloride (HY-17416). Guanfacine hydrochloride is an orally active noradrenergic α2A agonist and has high selective for the α2A receptor subtype. Guanfacine has effects in producing hypotension and sedation. Guanfacine can be used for the research of a variety of prefrontal cortex (PFC) cognitive disorders, including tourette's syndrome and attention deficit hyperactivity disorder (ADHD) .
|
-
- HY-12583
-
A-366
2 Publications Verification
|
Histone Methyltransferase
Epigenetic Reader Domain
|
Cancer
|
|
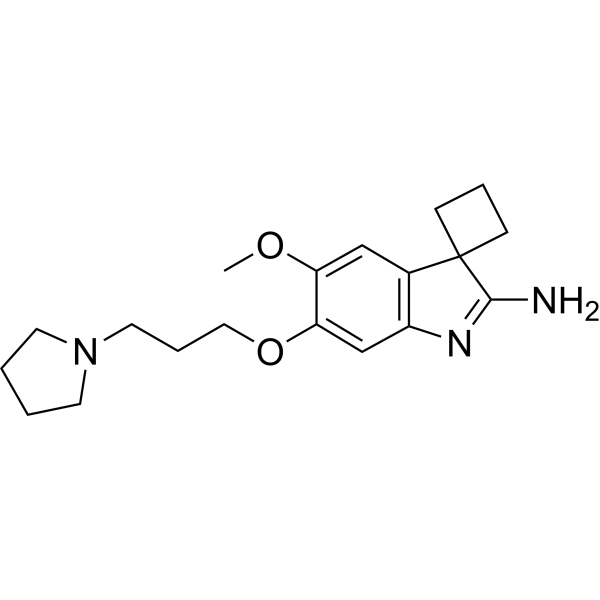
A-366 is a potent, highly selective, peptide-competitive histone methyltransferase G9a inhibitor with IC50s of 3.3 and 38 nM for G9a and GLP (EHMT1), respectively. A-366 shows >1000-fold selectivity over 21 other methyltransferases. A-366 is also a potent, nanomolar inhibitor of the Spindlin1-H3K4me3-interaction (IC50=182.6 nM). A-366 displays high affinity at human histamine H3 receptor (Ki=17 nM) and shows subtype selectivity among subsets of the histaminergic and dopaminergic receptor families .
|
-
- HY-12288
-
|
RPC-1063
|
LPL Receptor
|
Neurological Disease
Inflammation/Immunology
|
|
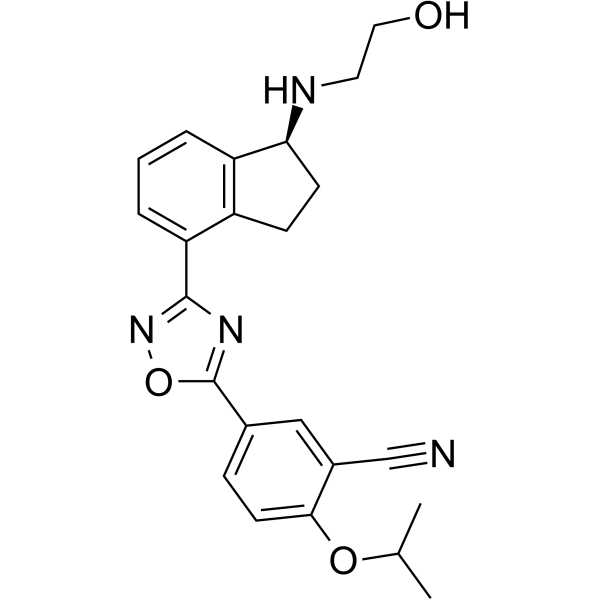
Ozanimod (RPC-1063), a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity selectively to S1P receptor subtypes 1 (S1P1) and 5 (S1P5). Ozanimod has modulate effect for hS1P1 and hS1P5 receptor with EC50s of 1.03 nM and 8.6 nM, respectively. Ozanimod can be used for the research of relapsing multiple sclerosis (MS) .
|
-
- HY-P1220
-
|
|
Sodium Channel
|
Neurological Disease
|
|
Huwentoxin-IV is a potent and selective sodium channel blocker, inhibits neuronal Nav1.7, Nav1.2, Nav1.3 and Nav1.4 with IC50s of 26, 150, 338 and 400 nM, respectively. Huwentoxin-IV preferentially blocks peripheral nerve subtype Nav1.7 by binding neurotoxin receptor site 4. Huwentoxin-IV has analgesic effects on animal models of inflammatory and neuropathic pain .
|
-
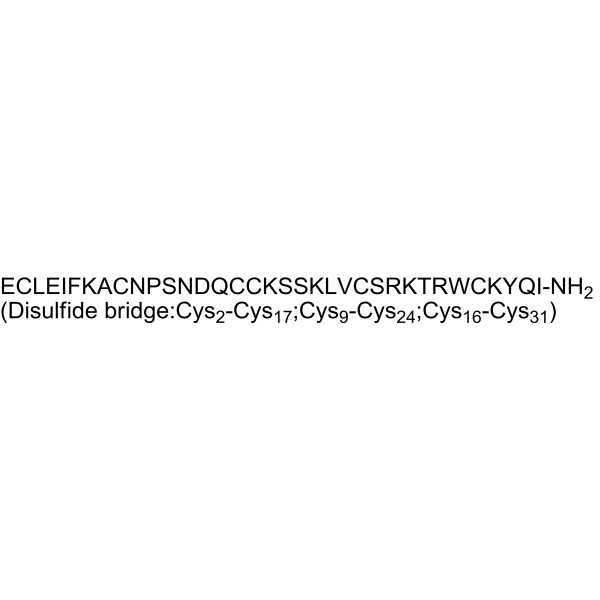
- HY-P1220A
-
|
|
Sodium Channel
|
Neurological Disease
|
|
Huwentoxin-IV TFA is a potent and selective sodium channel blocker, inhibits neuronal Nav1.7, Nav1.2, Nav1.3 and Nav1.4 with IC50s of 26, 150, 338 and 400 nM, respectively. Huwentoxin-IV TFA preferentially blocks peripheral nerve subtype Nav1.7 by binding neurotoxin receptor site 4. Huwentoxin-IV TFA has analgesic effects on animal models of inflammatory and neuropathic pain .
|
-

- HY-12288A
-
|
RPC-1063 hydrochloride
|
LPL Receptor
|
Neurological Disease
Inflammation/Immunology
|
|
Ozanimod (RPC-1063) hydrochloride, a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity selectively to S1P receptor subtypes 1 (S1P1) and 5 (S1P5). Ozanimod hydrochloride has modulate effect for hS1P1 and hS1P5 receptor with EC50s of 1.03 nM and 8.6 nM, respectively. Ozanimod hydrochloride can be used for the research of relapsing multiple sclerosis (MS) .
|
-
- HY-141547
-
|
|
Sodium Channel
Cytochrome P450
|
Inflammation/Immunology
|
|
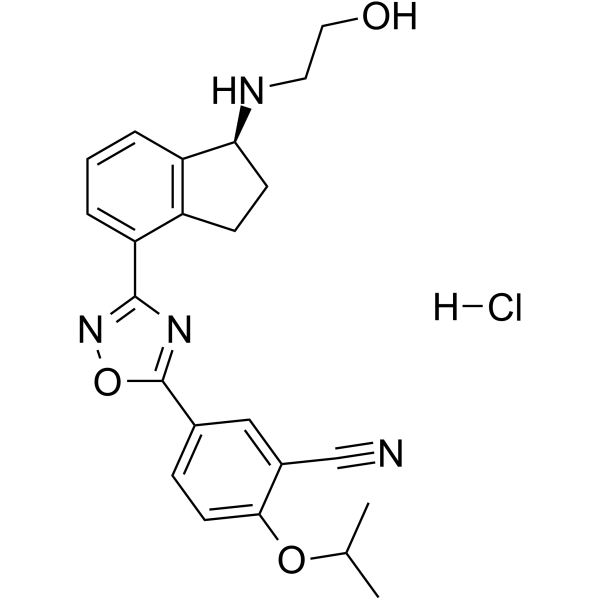
Nav1.7-IN-8 is a potent blockage of NaV1.7 with high selectivity for the inhibition of NaV1.7 over the subtypes hNaV1.1 and hNaV1.5. Nav1.7-IN-8 inhibits CYP2C9 and CYP3A4 with an IC50 of 0.17 μM and 0.077 μM, respectively. Nav1.7-IN-8 displays significant analgesic effects in rodent models of acute and inflammatory pain .
|
-
- HY-B1562
-
|
(±)-Bopindolol
|
|
|
|
Bopindolol ((±)-Bopindolol) is an orally active antagonist of β-adrenoceptors (ARs) with partial agonist activity. Bopindolol is non-selective for β1- and β2-ARs and has low affinity for β3-AR subtype. Bopindolol has intrinsic sympathomimetic as well as membrane stabilizing actions, inhibits renin secretion, and interacts with 5-HT receptors. Bopindolol is a proagent of Pindolol (HY-B0982). Bopindolol can be used for essential and renovascular hypertension research.
|
-
- HY-B1562C
-
|
(±)-Bopindolol fumarate
|
Adrenergic Receptor
Renin
5-HT Receptor
|
Cardiovascular Disease
Neurological Disease
|
|
Bopindolol ((±)-Bopindolol) fumarate is an orally active antagonist of β-adrenoceptors (ARs) with partial agonist activity. Bopindolol fumarate is non-selective for β1- and β2-ARs and has low affinity for β3-AR subtype. Bopindolol fumarate has intrinsic sympathomimetic as well as membrane stabilizing actions, inhibits renin secretion, and interacts with 5-HT receptors. Bopindolol fumarate is a proagent of Pindolol (HY-B0982). Bopindolol fumarate can be used for essential and renovascular hypertension research.
|
-
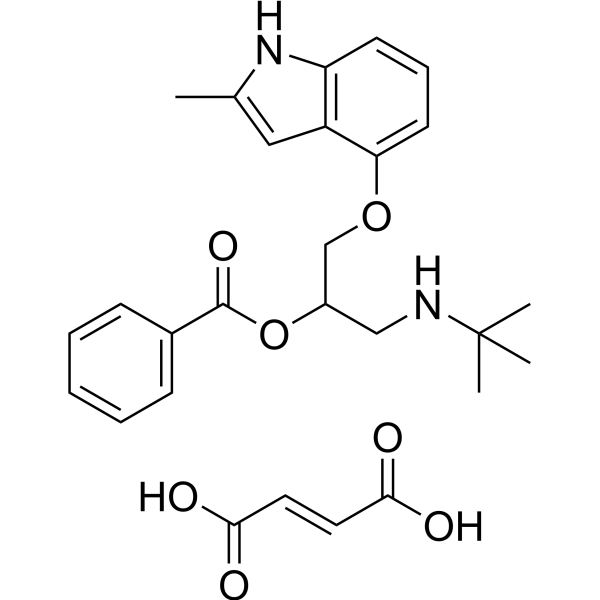
- HY-12709A
-
|
|
|
|
|
ARC 239 dihydrochloride is a selective α2B/2C adrenoceptor antagonist (pKd values are 5.95, 7.41 and 7.56 at α2A, α2B, and α2C receptors respectively). ARC 239 dihydrochloride binds to CHO cell membranes expressing human recombinant a2A-, a2B- or a2C-adrenoceptor subtypes with pKis of 5.6, 8.4, and 7.08, respectively .
|
-
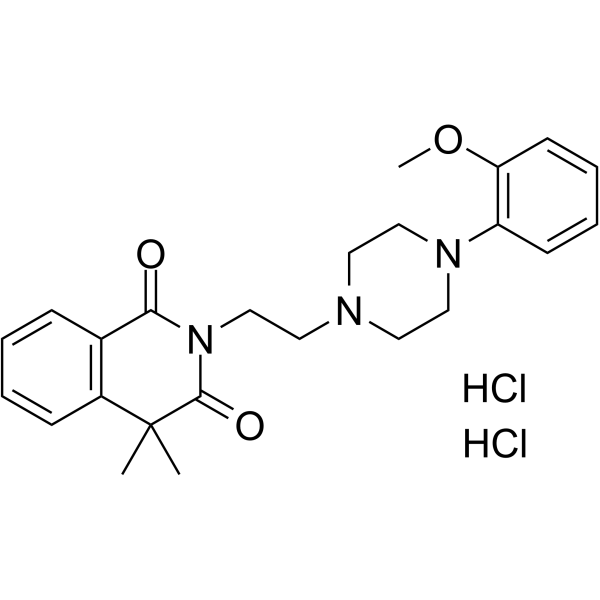
- HY-17606
-
|
ACT-334441
|
LPL Receptor
|
Inflammation/Immunology
|
|
Cenerimod (ACT-334441) is a potent, selective and orally active S1P1 receptor modulator, with an EC50 of 1 nM. Cenerimod shows more than 36 fold selctivity for hS1P1 over hS1P2, hS1P3, hS1P4, and hS1P5 receptor subtypes (EC50s=>10000, 228, 2134, and 36 nM, respectively). Cenerimod can attenuate murine experimental autoimmune encephalomyelitis (EAE) and murine sclerodermatous .
|
-
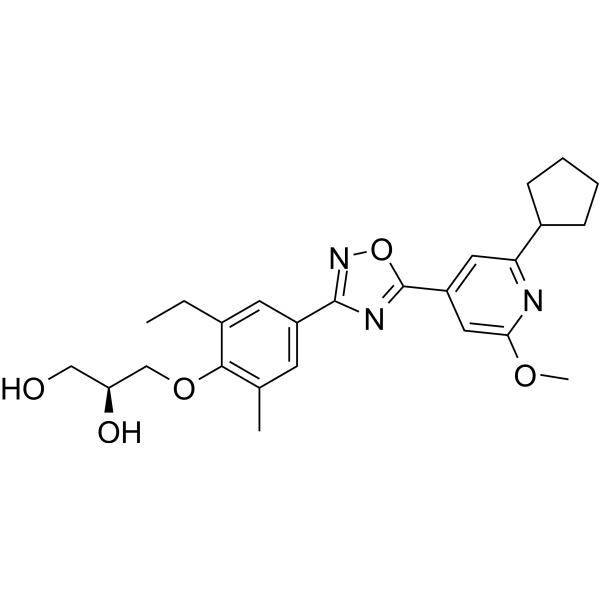
- HY-110105
-
|
|
Potassium Channel
|
Neurological Disease
|
|
NS8593 hydrochloride is a potent and selective small conductance Ca 2+-activated K + channels (SK channels) inhibitor. NS8593 hydrochloride reversibly inhibits SK3-mediated currents with a Kd value of 77 nM. NS8593 hydrochloride inhibits all the SK1-3 subtypes Ca 2+-dependently (Kds of 0.42, 0.60, and 0.73 μM, respectively, at 0.5 μM Ca 2+), and does not affect the Ca 2+-activated K + channels of intermediate and large conductance (hIK and hBK channels, respectively) .
|
-
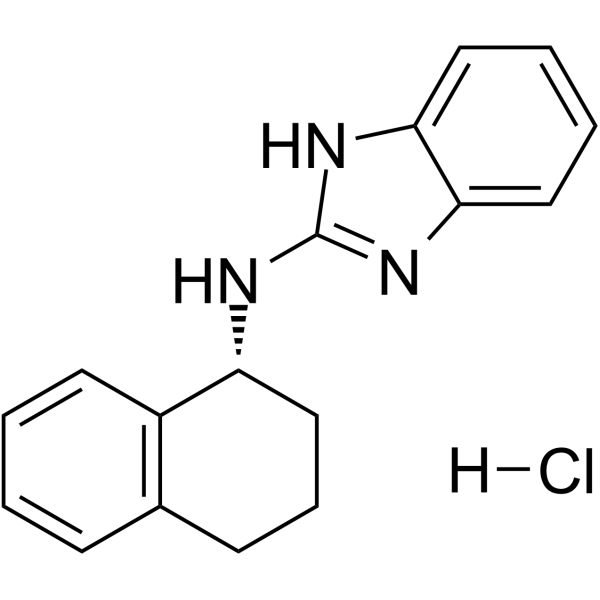
- HY-B1562B
-
|
(±)-Bopindolol (malonate)
|
Adrenergic Receptor
Renin
5-HT Receptor
|
Cardiovascular Disease
Neurological Disease
|
|
Bopindolol ((±)-Bopindolol) malonate is an orally active antagonist of β-adrenoceptors (ARs) with partial agonist activity. Bopindolol malonate is non-selective for β1- and β2-ARs and has low affinity for β3-AR subtype. Bopindolol malonate has intrinsic sympathomimetic as well as membrane stabilizing actions, inhibits renin secretion, and interacts with 5-HT receptors. Bopindolol malonate is a proagent of Pindolol (HY-B0982). Bopindolol malonate can be used for essential and renovascular hypertension research.
|
-
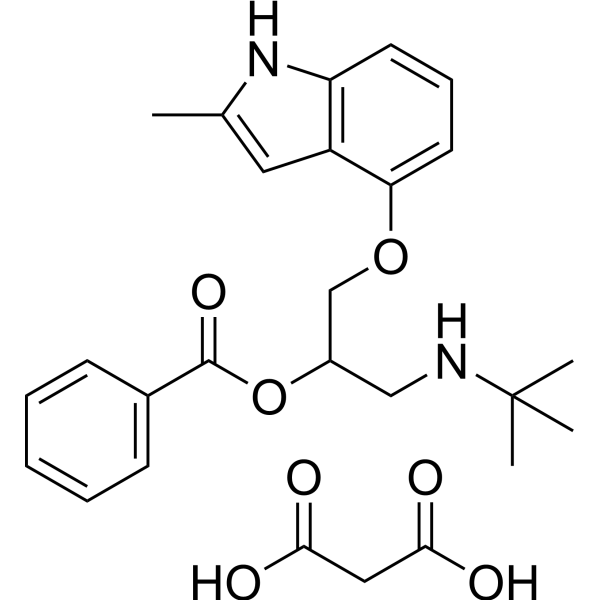
- HY-106954
-
|
Rec 15/2739; Recordati 15/2739; SB 216469
|
Adrenergic Receptor
|
Inflammation/Immunology
|
|
Upidosin (Rec 15/2739) is an α-1 adrenoceptor (α-1 AR) antagonist. Upidosin shows moderate selectivity for the α-1A AR subtype. Upidosin shows uroselectivity in urethra and prostate with a Kb value of 2-3 nM higher than in ear artery and aorta with a Kb value of 20-100 nM. Upidosin inhibits [3H]prazosin binding to cloned human α-1A adrenergic receptor. Upidosin can be used for the research of urethral obstruction .
|
-
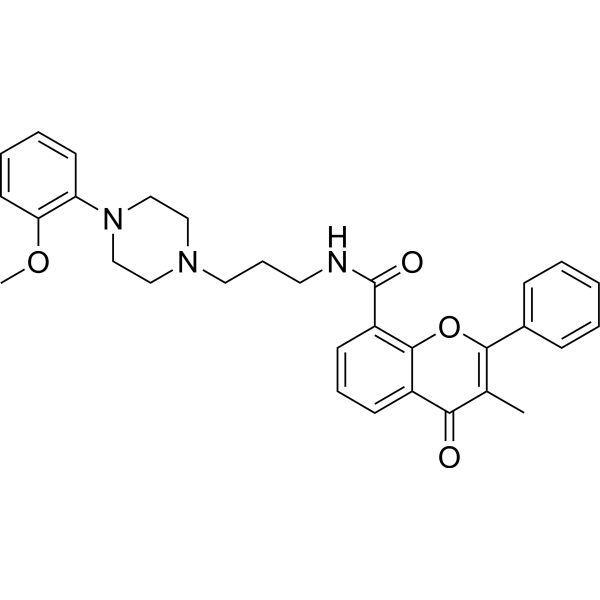
- HY-108501
-
|
|
Somatostatin Receptor
|
Neurological Disease
|
|
(1R,1'S,3'R/1R,1'R,3'S)-L-054,264 is a selective non-peptide human somatostatin receptor subtype 2 (sst2) agonist. (1R,1'S,3'R/1R,1'R,3'S)-L-054,264 can be used in the study of retinal neuromodulation .
|
-
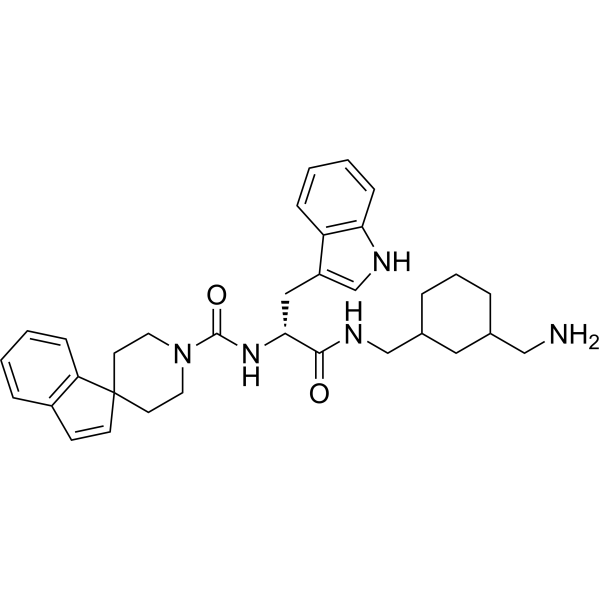
- HY-19210
-
|
|
Endothelin Receptor
|
Cardiovascular Disease
|
|
SB-209670 is an extremely potent and highly specific non-peptide, subnanomolar endothelin (ET) receptor antagonist. SB 209670 selectively inhibits binding of 125I-labeled ET-1 to cloned human ET receptor subtypes ETA and ETB (Ki=0.2 and 18 nM, respectively). SB 209670 produces a dose-dependent reduction in blood pressure in hypertensive rats, protects from ischemia-induced neuronal degeneration in a gerbil stroke model, and attenuates neointima formation following rat carotid artery balloon angioplasty .
|
-
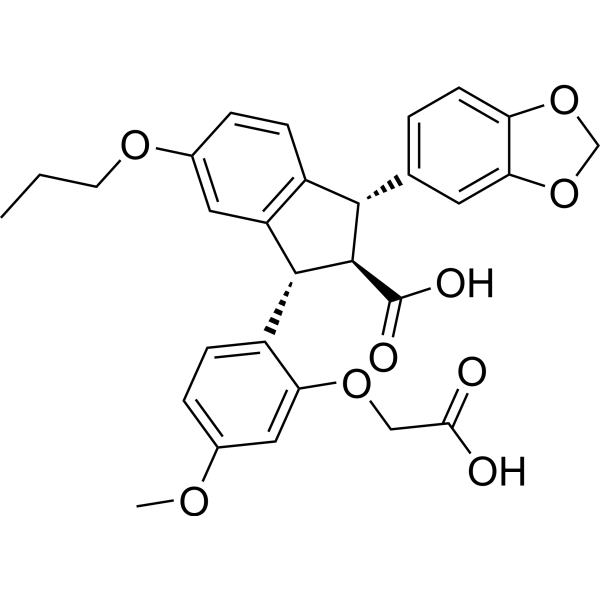
- HY-108495
-
|
ML248
|
LPL Receptor
|
Cardiovascular Disease
|
|
CYM50308 (ML248) is a potent, selective and high affinity sphingosine-1-phosphate receptor 4 (S1P4-R) agonist with an EC50 of 56 nM. CYM50308 displays 37-fold more selective for S1P4-R than S1P5-R. CYM50308 has no activity at S1P1-R, S1P2-R and S1P3-R subtypes at concentrations up to 25 μM .
|
-
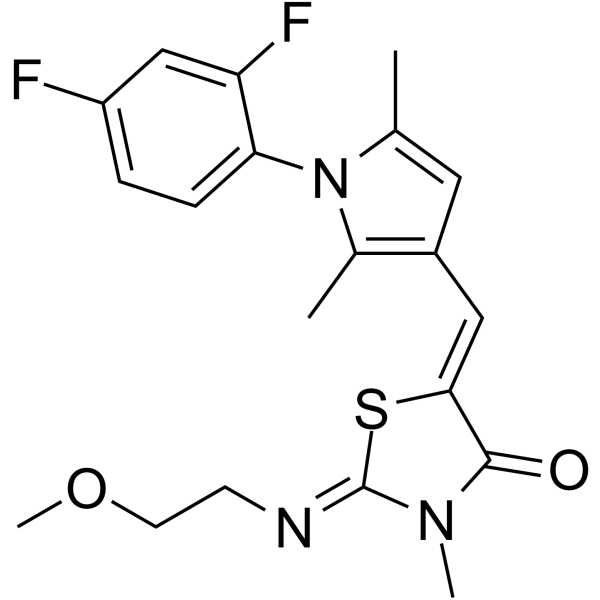
- HY-111052
-
|
|
GABA Receptor
Cytochrome P450
|
Neurological Disease
Inflammation/Immunology
|
|
AZD7325 is a potent and orally active partial selective PAM of GABAAα2 and Aα3 receptor (Ki=0.3 and 1.3 nM, respectively), and has less antagonistic efficacy at the Aα1 and Aα5 receptor subtypes . AZD7325 is a moderate CYP1A2 and a potent CYP3A4 inducer in vitro . AZD7325 has the potential for the investigation of anxiety and dravet syndrome . PAM: positive allosteric modulator.
|
-
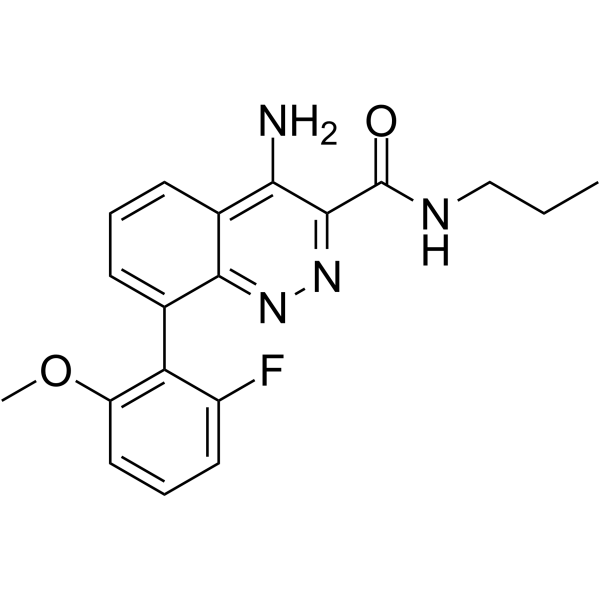
- HY-147319
-
|
|
Others
|
Neurological Disease
|
|
RTI-7470-44 is a potent, selective and blood-brain barrier (BBB) penetrant human trace amine-associated receptor subtype 1 (hTAAR1) antagonist with an IC50 value of 8.4 nM and a Ki value of 0.3 nM. RTI-7470-44 has moderate metabolic stability, and a favorable preliminary off-target profile. RTI-7470-44 can increase the spontaneous firing rate of mouse ventral tegmental area (VTA) dopaminergic neurons. RTI-7470-44 can be used for researching schizophrenia, agent addiction, and Parkinson’s disease (PD) .
|
-
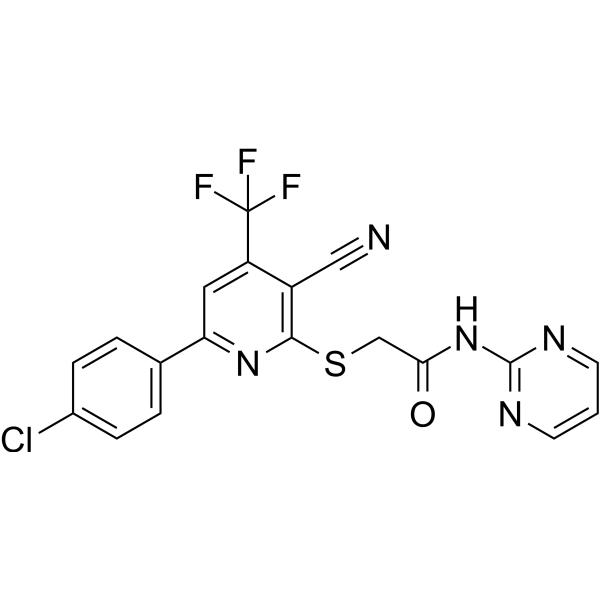
- HY-W009009
-
|
|
GABA Receptor
|
Neurological Disease
|
|
L-838417 is a selective partial agonist at the α2, α3 and α5 subtypes of the GABAA receptor and an antagonist at the α1, with binding Ki values of 0.79 nM, 0.67 nM, 1.67 nM, 267 nM, 2.25 nM and 2183 nM for α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, α5β3γ2 and α6β3γ2 .
|
-
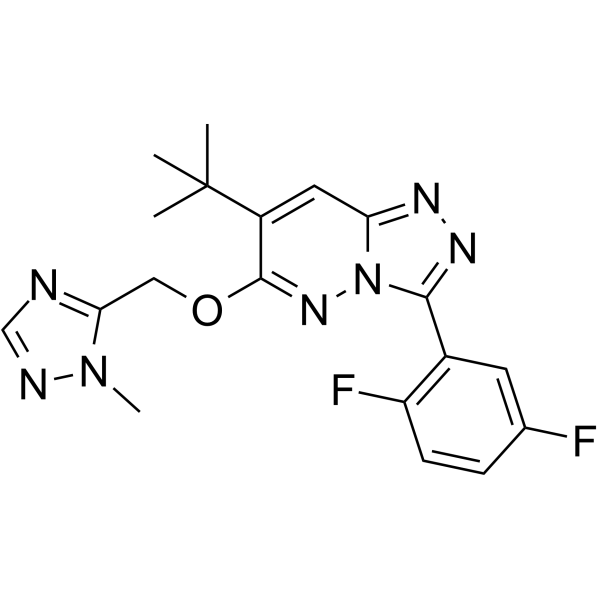
- HY-125777A
-
|
|
nAChR
|
Neurological Disease
|
|
α-Conotoxin Vc1.1 TFA is a disulfide-bonded peptide isolated from Conus victoriae and is a selective nAChR antagonist. α-Conotoxin Vc1.1 TFA inhibits α3α5β2, α3β2 and α3β4 with IC50s of 7.2 μM, 7.3 μM and 4.2 μM, respectively, and has less inhibitory effect on other nAChR subtypes. α-Conotoxin Vc1.1 TFA has the potential for neuropathic pain reserach .
|
-

- HY-138879B
-
|
(1S,5R)-CP-601927
|
nAChR
|
Neurological Disease
|
|
CP-601932 ((1S,5R)-CP-601927) is a high-affinity partial agonist at α3β4 nAChR (Ki=21 nM; EC50=~ 3 μM). CP-601932 has the same high-binding affinity at α4β2 nAChR (Ki=21 nM) and an order of magnitude lower affinity for α6 and α7 nAChR subtypes. CP-601932 selectively decreases ethanol but not sucrose consumption and operant self-administration following long-term exposure. CP-601932 can penetrate the CNS .
|
-
- HY-103216A
-
|
|
Adrenergic Receptor
|
Neurological Disease
|
|
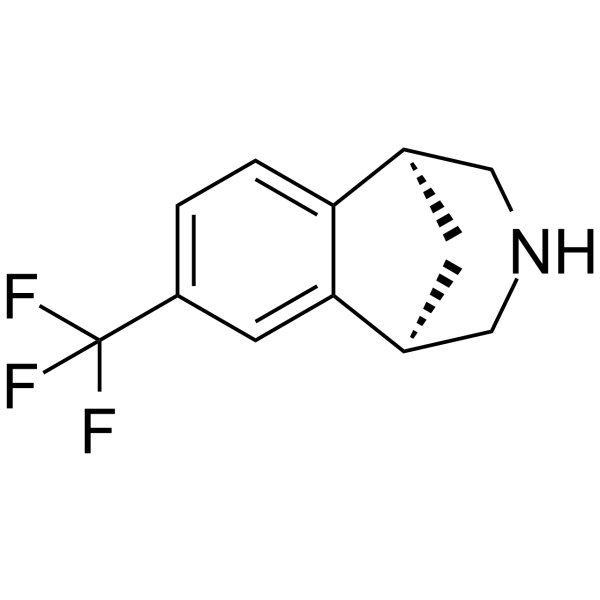
RWJ-52353 hydrochloride is an orally potent, highly selective α2D adrenergic receptor agonist (Ki: 1.5 nM) with potential analgesic effects. RWJ-52353 hydrochloride demonstrated analgesic activity in abdominal tests in rats and mice, and improved agitation in mice in the hot plate test and tail flick test. RWJ-52353 hydrochloride also regulates the organic cation transporter (OCT) subtype, inhibiting rOCT1 and rOCT2 with IC50s of 100 μM and 20 μM respectively; it also activates rOCT3, affecting [3H]-1- in cells. Methyl-4-phenylpyridinium ([3H]MPP) transport .
|
-
- HY-120874
-
|
|
GABA Receptor
|
Neurological Disease
|
|
PF-06372865 is an orally active, α2/α3/α5 subtype-selective GABAA positive allosteric modulator (PAM). PF-06372865 is a high affinity ligand at GABAA receptors containing α1/α2/α3/α5 subunits (Kis of 2.9 nM, 21 nM, 134 nM for α2, α1 PAM, α2 PAM, respectively), with low affinity for α4/α6 subunits. PF-06372865 can across the blood-brain barrier (BBB). PF-06372865 has anxiolytic activity and has the potential for epilepsy .
|
-
- HY-120973
-
|
|
Biochemical Assay Reagents
|
Others
|
|
(R)-Butaprost (free acid). Butaprost is a structural analog of prostaglandin E2 (PGE2) with good selectivity for the EP2 receptor subtype. Butaprost is frequently used pharmacologically to define the expression profile of EP receptors in various human and animal tissues and cells. Gardiner caused serious confusion about the structure of butaprost in 1986 when he reported that the epimer of butaprost showing this selective activity was the C-16 (R)-epimer ( See reference 2 and notes). To increase the binding affinity of (R)-butaprost to prostaglandin receptors, we removed the methyl ester of (R)-butaprost and recreated the native C-1 carboxylic acid. Prostaglandin free acids typically bind their cognate receptors with 10 to 100-fold higher affinity than the corresponding ester derivatives. The pharmacology of (R)-butaprost has not been carefully studied, but it is generally considered to be the less active C-16 epimer. (Note: In the 1986 Gardiner paper in the British Journal of Pharmacology, butaprost appears on page 46 under the designation TR 4979. The structure drawn is incorrect because the authors use and refer to the more active C - The 16 epimer, which is actually 16(S). The structure on page 46 shows the structure as 16(R). It was not until the late 1990s that careful studies in the United States and Japan correctly determined the actual structure of C-16 The type is 16(S) in a compound called butaprost.)
|
-
-
-
HY-L159
-
|
|
1468 compounds
|
|
Agonistic drugs activate or stimulate their receptors, triggering responses that increase or decrease cell activity. The highly selective activators can act on specific biological or molecular targets, while non-selective activators may interfere with multiple targets or targets simultaneously. The highly selective activators reduce the likelihood of these non-specific effects by targeting specific targets, making research more precise and reliable. The Highly Selective Activators Library contains 1468 compounds, covering multiple targets and subtypes, such as GPCR protein family, Ion channel, multiple kinases, etc. The Highly Selective Activators Library is an effective tool for screening different phenotypes.
|
-
-
HY-L158
-
|
|
4653 compounds
|
|
According to reports, most known kinase inhibitors exert their effects through competitive binding in highly conserved ATP pockets. Although genetic techniques such as RNA interference can inactivate specific genes, most kinases are multi domain proteins, each of which has an independent function. Highly selective inhibitors have higher efficiency than non-selective inhibitors, and the selectivity to the target is at least 100 times higher. Therefore, ensuring the validation of targets with the most selective inhibitors is crucial for a more thorough understanding of the pharmacology of the kinase field. The Highly Selective Inhibitors Library contains 4653 compounds, covering multiple targets and subtypes, such as GPCR protein family, Ion channel, multiple kinases, etc. The Highly Selective Inhibitors Library is an effective tool for screening different phenotypes
|
-
-
HY-L170
-
|
|
174 compounds
|
|
An emerging drug design method is based on the secondary binding site effect, where small molecule drugs are designed to bind to secondary binding sites on target biomolecules rather than primary orthomorphic sites. Successful potential drugs (known as allosteric modulators) will be able to bind to allosteric sites and remotely alter (or modify) the conformation of the main orthosteric binding sites of biological targets. Allosteric modulators (AMs) are ligands of proteins that act through binding sites different from natural (orthosteric) ligand sites. AMs are relatively small, more lipophilic, and more rigid compounds. The binding efficacy of AMs with their targets is often slightly lower. AMs are divided into positive AMs (PAMs) and negative AMs (NAMs). AMs are ideal drug targets because they can fine-tune receptor activity while preserving the spatial and temporal signal transduction characteristics of endogenous ligands, resulting in fewer targeted side effects, improved subtype selectivity, and better promotion of biased signal transduction than normal ligands.
MCE designs a unique collection of 174 small allosteric modulators. It is a good tool to be used for research on metabolize, cancer and other diseases.
|
| Cat. No. |
Product Name |
Type |
-
- HY-120973
-
|
|
Biochemical Assay Reagents
|
|
(R)-Butaprost (free acid). Butaprost is a structural analog of prostaglandin E2 (PGE2) with good selectivity for the EP2 receptor subtype. Butaprost is frequently used pharmacologically to define the expression profile of EP receptors in various human and animal tissues and cells. Gardiner caused serious confusion about the structure of butaprost in 1986 when he reported that the epimer of butaprost showing this selective activity was the C-16 (R)-epimer ( See reference 2 and notes). To increase the binding affinity of (R)-butaprost to prostaglandin receptors, we removed the methyl ester of (R)-butaprost and recreated the native C-1 carboxylic acid. Prostaglandin free acids typically bind their cognate receptors with 10 to 100-fold higher affinity than the corresponding ester derivatives. The pharmacology of (R)-butaprost has not been carefully studied, but it is generally considered to be the less active C-16 epimer. (Note: In the 1986 Gardiner paper in the British Journal of Pharmacology, butaprost appears on page 46 under the designation TR 4979. The structure drawn is incorrect because the authors use and refer to the more active C - The 16 epimer, which is actually 16(S). The structure on page 46 shows the structure as 16(R). It was not until the late 1990s that careful studies in the United States and Japan correctly determined the actual structure of C-16 The type is 16(S) in a compound called butaprost.)
|
| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-P1221A
-
|
|
Sodium Channel
|
Neurological Disease
|
|
ProTx II TFA is a selective blocker of Nav1.7 sodium channels with an IC50 of 0.3 nM, and is at least 100-fold selective for Nav1.7 over other sodium channel subtypes. ProTx-II inhibits sodium channels by decreasing channel conductance and shifting activation to more positive potentials and blocks action potential propagation in nociceptors .
|
-
- HY-P1219
-
|
β-TRTX-Cj1α
|
Sodium Channel
|
Neurological Disease
|
|
Jingzhaotoxin-III is a potent and selective blocker of Nav1.5 channels, with an IC50 of 348 nM, and shows no effect on other sodium channel isoforms. Jingzhaotoxin-III can selectively inhibit the activation of cardiac sodium channel but not neuronal subtypes, and hopefully represents an important ligand for discriminating cardiac VGSC subtype .
|
-
- HY-P5158
-
|
|
Adrenergic Receptor
|
Others
|
|
Conopeptide rho-TIA is a peptide derived from the venom contained in the predatory sea snail Conus tulipa, has highly selective and noncompetitive inhibitor at human α1B-Adrenergic Receptor. Conopeptide rho-TIA acts a competitive inhibitor at human α1A-Adrenergic Receptor and α1D-Adrenergic Receptor. Conopeptide rho-TIA binds to each subtype and may provide useful information for the development of novel α1-Adrenergic Receptor subtype-selective drugs .
|
-
- HY-P5172
-
|
|
Sodium Channel
|
Neurological Disease
|
|
MitTx-alpha is a subunit of MitTx. MitTx is a potent, persistent, and selective agonist for acid-sensing ion channels (ASICs). MitTx is highly selective for the ASIC1 subtype at neutral pH; under more acidic conditions (pH<6.5), MitTx massively potentiates (>100-fold) proton-evoked activation of ASIC2a channels .
|
-
- HY-P1221
-
|
|
Sodium Channel
|
Neurological Disease
|
|
ProTx II is a selective blocker of Nav1.7 sodium channels with an IC50 of 0.3 nM, and is at least 100-fold selective for Nav1.7 over other sodium channel subtypes. ProTx-II inhibits sodium channels by decreasing channel conductance and shifting activation to more positive potentials and blocks action potential propagation in nociceptors .
|
-
- HY-P5010
-
|
|
Vasopressin Receptor
|
Cardiovascular Disease
|
|
(D-Arg8)-Inotocin is a potent, selective and competitive antagonist of vasopressin receptor (V1aR), with a Ki of 1.3 nM. (D-Arg8)-Inotocin shows more than 3000-fold selectivity for the human V1aR over the other three subtypes, OTR, V1bR and V2R .
|
-
- HY-P3676
-
|
|
Neuropeptide Y Receptor
|
Neurological Disease
|
|
Neuropeptide Y (3-36) (porcine) is an agonist of neuropeptide Y (NPY) receptor subtype Y2, and stimulates feeding in rats. Neuropeptide Y (3-36) (porcine) is a highly Y2 selective ligand compared with nselective Y1/Y2 receptor ligand, Neuropeptide Y 1-36 .
|
-
- HY-P1220
-
|
|
Sodium Channel
|
Neurological Disease
|
|
Huwentoxin-IV is a potent and selective sodium channel blocker, inhibits neuronal Nav1.7, Nav1.2, Nav1.3 and Nav1.4 with IC50s of 26, 150, 338 and 400 nM, respectively. Huwentoxin-IV preferentially blocks peripheral nerve subtype Nav1.7 by binding neurotoxin receptor site 4. Huwentoxin-IV has analgesic effects on animal models of inflammatory and neuropathic pain .
|
-
- HY-P1220A
-
|
|
Sodium Channel
|
Neurological Disease
|
|
Huwentoxin-IV TFA is a potent and selective sodium channel blocker, inhibits neuronal Nav1.7, Nav1.2, Nav1.3 and Nav1.4 with IC50s of 26, 150, 338 and 400 nM, respectively. Huwentoxin-IV TFA preferentially blocks peripheral nerve subtype Nav1.7 by binding neurotoxin receptor site 4. Huwentoxin-IV TFA has analgesic effects on animal models of inflammatory and neuropathic pain .
|
-
- HY-125777A
-
|
|
nAChR
|
Neurological Disease
|
|
α-Conotoxin Vc1.1 TFA is a disulfide-bonded peptide isolated from Conus victoriae and is a selective nAChR antagonist. α-Conotoxin Vc1.1 TFA inhibits α3α5β2, α3β2 and α3β4 with IC50s of 7.2 μM, 7.3 μM and 4.2 μM, respectively, and has less inhibitory effect on other nAChR subtypes. α-Conotoxin Vc1.1 TFA has the potential for neuropathic pain reserach .
|
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
| Cat. No. |
Product Name |
Chemical Structure |
-
- HY-15831
-
|
|
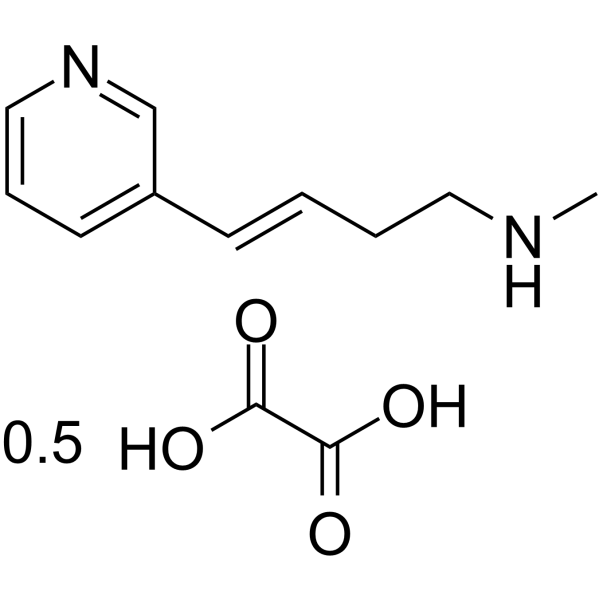
|
L-838417-d9 is the deuterium labeled L-838417. L-838417 is a subtype-selective GABAA positive allosteric modulator, acting as a partial agonist at α2, α3 and α5 subtypes[1].
|
-
-
- HY-B0197AS
-
|
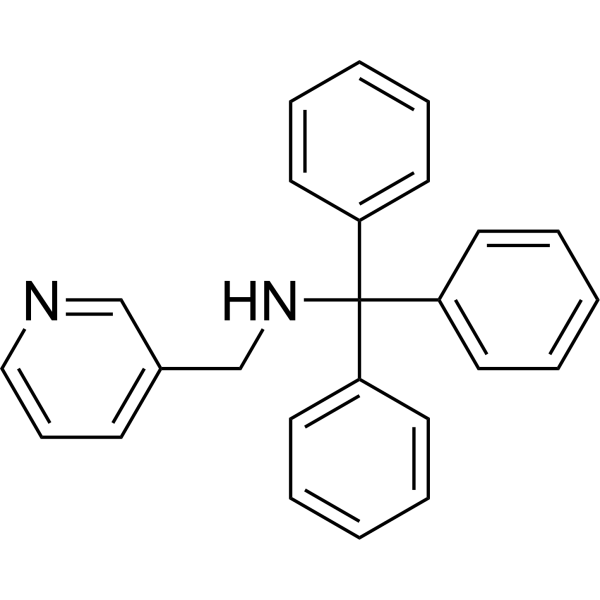
|
|
Naratriptan-d3 (hydrochloride) is the deuterium labeled Naratriptan, which is a selective 5-HT1 receptor subtype agonist.
|
-
-
- HY-B0197S
-
|
|
|
Naratriptan-d3 is the deuterium labeled Naratriptan[1]. Naratriptan is a selective 5-HT1 receptor subtype agonist[2].
|
-
-
- HY-B0374S
-
|
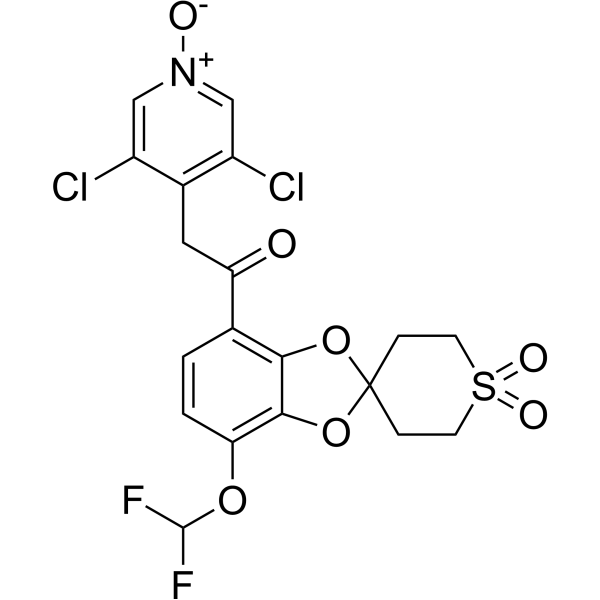
|
|
Moxonidine-d4 (BDF5895-d4) is the deuterium labeled Moxonidine. Moxonidine(BDF5895) is a selective agonist at the imidazoline receptor subtype 1, used as antihypertensive agent[1][2].
|
-
-
- HY-B0391S1
-
|
|
|
Naftopidil-d5 is deuterium labeled Naftopidil. Naftopidil (KT-611) is is a selective alpha1-adrenoceptor antagonist, with Kis of 3.7 nM, 20 nM and 1.2 nM for the cloned human α1a-, α1b- and α1d-adrenoceptor subtypes, respectively. Naftopidil has antiproliferative effects. Naftopidil can be used for the research of prostate hyperplasia[1][2].
|
-
-
- HY-17416AS
-
|
|
|
Guanfacine- 13C, 15N3 is the 13C and 15N labeled Guanfacine[1]. Guanfacine is an orally active noradrenergic α2A agonist and has high selective for the α2A receptor subtype. Guanfacine has effects in producing hypotension and sedation. Guanfacine can be used for the research of a variety of prefrontal cortex (PFC) cognitive disorders, including tourette's syndrome and attention deficit hyperactivity disorder (ADHD)[2][3][4].
|
-
-
- HY-17416S2
-
|
|
|
Guanfacine- 13C,d5 hydrochloride is the deuterium and 13C labeled Guanfacine hydrochloride (HY-17416). Guanfacine hydrochloride is an orally active noradrenergic α2A agonist and has high selective for the α2A receptor subtype. Guanfacine has effects in producing hypotension and sedation. Guanfacine can be used for the research of a variety of prefrontal cortex (PFC) cognitive disorders, including tourette's syndrome and attention deficit hyperactivity disorder (ADHD) .
|
-
| Cat. No. |
Product Name |
|
Classification |
-
- HY-123249
-
|
|
|
Alkynes
|
|
HZ166 is a GABAA receptor subtype-selective benzodiazepine site agonist with preferential activity at α2- and α3-GABAA receptors. HZ166 shows anti-hyperalgesic effects . HZ166 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent: